





Label-Free Quantitative Service
- Optimize Sample Preparation: Improve protein extraction methods, such as using different lysis buffers and concentration techniques, to enhance low-abundance protein recovery.
- Enhance Mass Spectrometry Sensitivity: Utilize high-resolution mass spectrometers (e.g., Orbitrap) with high-sensitivity acquisition modes to improve the detection of low-abundance peptides.
- Optimize Liquid Chromatography Separation: Select the appropriate chromatography column and separation conditions to maximize peptide separation, reducing co-elution interference and improving the identification of low-abundance peptides.
- Refine Data Analysis Strategies: Use advanced data processing algorithms (such as strategies to maximize spectral matching and peptide quantification), combined with various normalization methods (such as internal reference protein normalization), to reduce systematic errors.
- Internal Standards: Incorporate stable internal standards, such as heavily labeled reference materials or known-abundance internal proteins, to normalize samples and reduce technical variability.
- Optimized Data Processing Algorithms: Use advanced data processing tools (e.g., MaxQuant, Proteome Discoverer) along with appropriate quality filtering criteria (e.g., removing low signal-to-noise ratio data) to improve quantitative accuracy.
- Multiple Normalization Approaches: In order to reduce the batch effect between samples, a variety of normalization strategies can be used, such as TIC (total ion current) normalization, internal reference protein normalization, etc., to ensure comparability between different samples.
- Data Correction: Use machine learning algorithms for data correction, taking into account sample complexity, technical variation, etc., to further improve the reliability of quantitative data.
Label-Free Quantitative Service allows for direct comparison of protein expression levels across different samples without the need for isotope labeling, and is suitable for multi-omics research, disease mechanism analysis, and protein interaction network studies. Label-Free Quantitative (LFQ) is a liquid chromatography-mass spectrometry (LC-MS/MS)-based quantitative proteomics approach that measures peptide signal intensity or spectral counting to assess the relative abundance of proteins in a sample. Compared to isotope labeling-based quantification, LFQ offers advantages such as eliminating isotope labeling requirements, reducing experimental costs, increasing throughput, and simplifying sample preparation, making it widely used in large-scale proteomics studies. Leveraging an advanced mass spectrometry platform and extensive technical expertise, MtoZ Biolabs provides Label-Free Quantitative Service to support quantitative proteomics research, helping researchers obtain highly reliable and accurate protein quantification data.
Analysis Workflow
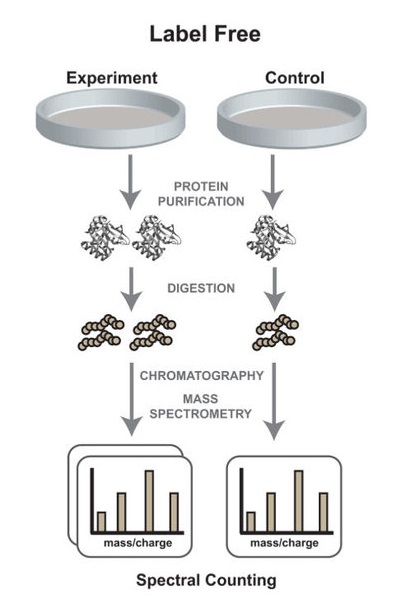
Miah S. et al. Molecular BioSystems. 2016.
1. Sample Preparation
Extract proteins and perform enzymatic digestion to generate peptide fragments.
2. LC-MS/MS Analysis
Use nano liquid chromatography (nano-LC) coupled with high-resolution mass spectrometry (e.g., Orbitrap) for peptide separation and detection. Acquire MS1 (peptide ion intensity) and MS2 (fragmentation spectra) data.
3. Data Processing
Perform data analysis and protein identification using software such as MaxQuant and databases like Uniprot and SwissProt. Apply LFQ algorithms to calculate relative protein abundance, normalize data, and remove background noise.
4. Biological Analysis
Statistical analysis (e.g., t-test, ANOVA, PCA) to identify differentially expressed proteins.
Enrichment analysis (e.g., GO, KEGG) to explore functional pathways.
Protein-protein interaction (PPI) network analysis to uncover biological regulatory mechanisms.
Service Advantages
1. Advanced Analysis Platform: MtoZ Biolabs established an advanced Label-Free Quantitative Service platform, guaranteeing reliable, fast, and highly accurate analysis service.
2. One-Time-Charge: Our pricing is transparent, no hidden fees or additional costs.
3. High-Data-Quality: Deep data coverage with strict data quality control. AI-powered bioinformatics platform integrates all Label-Free Quantitative Service data, providing clients with a comprehensive data report.
4. High-throughput analysis for large-scale studies: Capable of analyzing a large number of samples in a single run, making it ideal for clinical research and large-scale screening.
5. Broad applicability across various sample types: Compatible with cells, tissues, plasma, urine, FFPE samples and more, without requiring additional labeling steps.
Applications
1. Biomarker Discovery
Compare Label-Free Quantitative proteomics data between healthy and pathological samples to identify disease-associated biomarkers.
2. Cell Signaling Pathway Research
Analyze dynamic protein expression changes in response to stimuli, revealing regulatory mechanisms in signaling pathways such as MAPK and PI3K/AKT.
3. Drug Action Mechanism and Target Discovery
Evaluate the effects of drug treatments on protein expression in cells or tissues to identify potential drug targets and resistance mechanisms.
4. Immunoproteomics
Investigate dynamic protein expression changes during immune responses, including cytokine regulation and antibody responses.
5. Microbial and Environmental Proteomics
Analyze protein expression in environmental microbes or symbiotic microbiota to uncover their impact on hosts or ecosystems.
FAQ
Q. How can the quantification accuracy of low-abundance proteins be improved?
LFQ technology is more accurate in quantifying high-abundance proteins, but the quantification of low-abundance proteins is usually limited by the sensitivity of mass spectrometry. To improve low-abundance protein quantification accuracy, the following strategies can be employed:
Q. How can reliable quantitative information be extracted from complex data?
To extract reliable quantitative information from complex mass spectrometry data, the following strategies can be employed:
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Case Study
This study conducted a label-free quantitative analysis of protein expression in the serum of patients with type 2 diabetes (T2DM) and healthy people, and identified a total of 1,074 proteins, of which 90 were significantly differentially expressed between T2DM patients and healthy controls, including 32 proteins that were first discovered to be associated with T2DM. Subsequently, the researchers used enzyme-linked immunosorbent assay (ELISA) to verify the candidate proteins in an independent validation cohort. The results of this study provide new potential biomarkers for the early diagnosis of T2DM, which will help improve the accuracy and timeliness of clinical diagnosis.
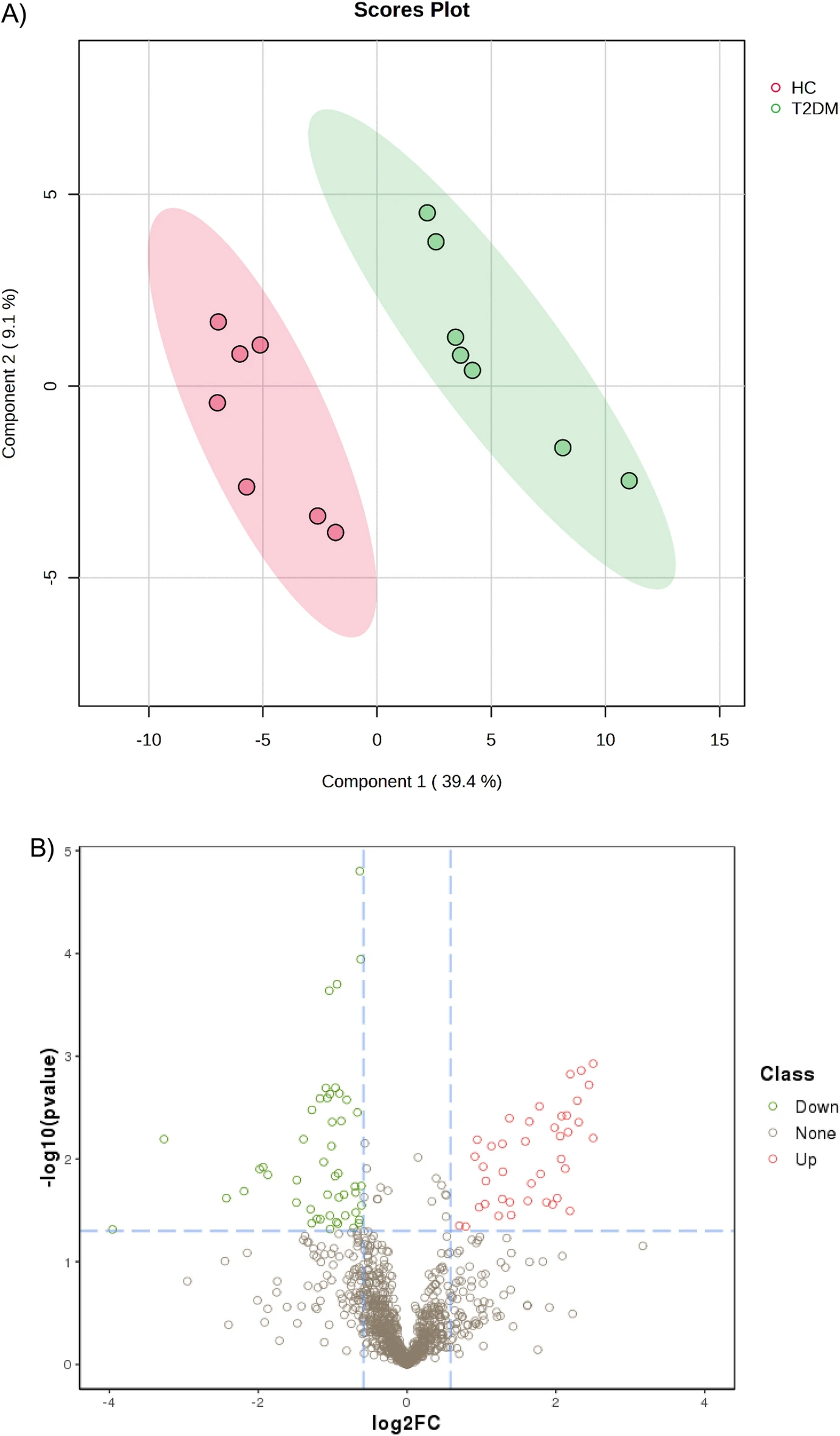
Nimer R M. et al. Sci Rep. 2023.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Quantitative Proteomics Service
Label-Free Quantitative Proteomics Service | Quantitative Phosphoproteomics
How to order?

