





IP-MS Protein Interactomics Analysis Service
- Ubiquitination-dependent interaction dynamics
- Composition and functional dissection of chromatin regulatory complexes
- Differential interactome analysis of tumor-associated protein mutants
- Comprehensive mapping of virus–host protein interactions
Protein-protein interactions (PPIs) orchestrate virtually every cellular process, from signal transduction and transcriptional regulation to structural organization and metabolic coordination. Mapping these interactions is essential for understanding protein function, disease mechanisms, and identifying novel therapeutic targets.
Immunoprecipitation–Mass Spectrometry (IP-MS) integrates the targeted enrichment capabilities of co-immunoprecipitation (Co-IP) with the high-resolution identification power of liquid chromatography–tandem mass spectrometry (LC-MS/MS). This powerful technique enables detailed characterization of protein complex composition and dynamic interaction networks. Unlike other interaction-mapping approaches such as yeast two-hybrid (Y2H), BioID, or cross-linking mass spectrometry (XL-MS), IP-MS offers the distinct advantage of capturing protein interactions under endogenous or near-physiological conditions. It is suitable for both confirming known interactions and uncovering novel regulatory pathways, making IP-MS a cornerstone in mapping eukaryotic protein interactomes.
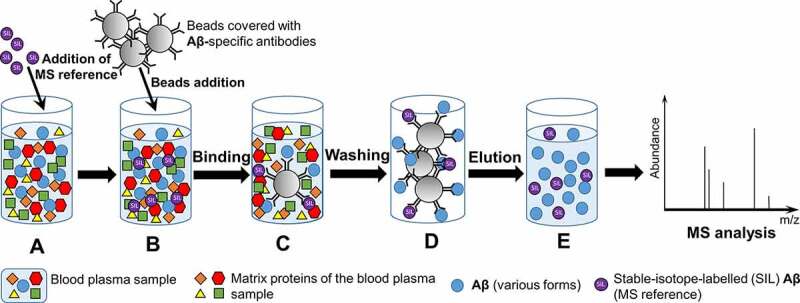
Kulichikhin, K. Y. et al. Prion. 2021.
Figure1. Principle of the ImmunoPrecipitation Followed by Mass Spectrometry (IP-MS)
Service at MtoZ Biolabs
MtoZ Biolabs offers high-quality IP-MS interactomics analysis services by combining high-affinity antibody enrichment with advanced LC-MS/MS platforms, including the Thermo Fisher Orbitrap Fusion Lumos and Q Exactive HF. Target proteins and their interaction partners are enriched using high-affinity antibodies or epitope tags, followed by identification and quantification through high-resolution Orbitrap mass spectrometry. This enables accurate profiling of both stable and transient protein interactions across various sample types.
Our IP-MS interactomics analysis services supports comparative interaction analysis under different experimental conditions, such as drug treatment or mutant screening. In addition, we provide comprehensive data evaluation workflows, including CRAPome filtering and SAINT scoring, making this service well-suited for pathway analysis, functional validation of mutations, and the construction of dynamic protein interaction maps.
Technical Principles
IP-MS integrates the target specificity of affinity purification with the throughput and resolution of mass spectrometry to map protein interaction networks within cells. This method relies on antibody- or tag-based enrichment strategies to capture the target protein and its associated complex members under mild lysis conditions. The enriched components are then enzymatically digested and analyzed by mass spectrometry to identify and quantify interacting proteins.
The general workflow includes the following steps: 1)non-denaturing cell lysis to extract soluble protein complexes; 2)immunoprecipitation using magnetic beads or agarose-conjugated antibodies; 3)enzymatic digestion of enriched components with trypsin; 4)LC-MS/MS analysis of resulting peptides; and 5)database searching and statistical filtering to identify high-confidence interactors.
To minimize background interference, experiments are typically designed with appropriate control groups and analyzed using tools such as the CRAPome database or SAINT algorithm for data filtering. Depending on the research objective, quantitative interaction proteomics can be performed using strategies such as TMT or SILAC, enabling comparative analyses across multiple conditions.
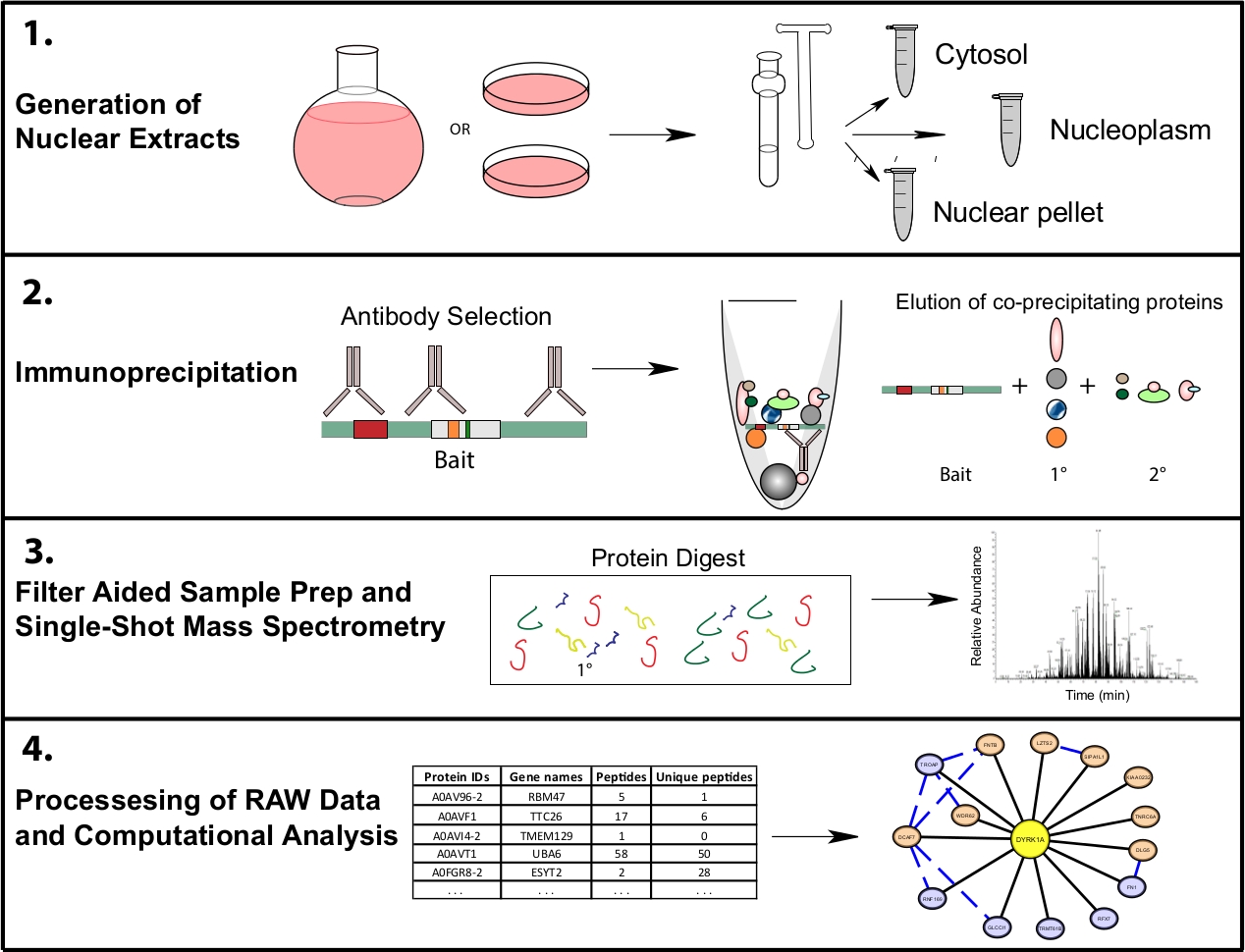
Guard, S. E. et al. J. Vis. Exp. 2019.
Figure 2. Representative Proteomics Workflow for Subcellular IP-MS
Service Advantages
1. Physiological relevance: Enables direct extraction from cells or tissues without the need for overexpression systems, thereby reducing the risk of false positives.
2. High specificity and resolution: Combines antibody-based affinity capture with mass spectrometry to enhance the confidence in complex composition identification.
3. Dynamic interaction compatibility: Supports experimental designs involving time-course studies, stimulus treatments, or genetic mutations to assess interaction changes.
4. Flexible quantification strategies: Compatible with label-free, SILAC, and TMT approaches to meet various experimental requirements.
5. Integrative downstream analysis: Results can be further analyzed using STRING, Cytoscape, IPA, and other tools for network modeling and pathway enrichment.
Applications
1.Reconstruction of signaling interaction networks
Identify upstream and downstream interaction partners regulated by ligand stimulation, drug treatment, or other condition changes.
2.Comparative analysis of wild-type and mutant protein interactions
Analyze interaction differences between wild-type and mutant proteins to uncover mechanisms of gain or loss of function.
3.Characterization of chromatin or transcriptional complexes
Applicable to the study of transcription factors, chromatin-modifying enzymes, and other regulatory protein complexes.
4.Detection of PTM-dependent interactions (e.g., ubiquitination, phosphorylation)
Investigate modification-dependent interactions involving kinases, E3 ligases, and reader proteins.
5.Mapping of virus–host protein interactions
Systematically reveal interaction profiles between viral proteins and host signaling components.
Case Study
Captured interaction changes associated with the ubiquitination status of target proteins, providing mechanistic insights into PTM-regulated signaling networks.
Zhou Y et al., Nature Chemical Biology, 2021
Elucidated the interaction architecture and functional roles of chromatin-associated protein complexes involved in transcriptional regulation.
Shan H et al., Molecular Cell, 2023
Compared wild-type and mutant protein interactomes in cancer models, revealing gain- and loss-of-function mechanisms linked to oncogenic mutations.
Karsli Uzunbas H et al., Cell Reports, 2022
Established a global protein interaction landscape between viral factors and host signaling proteins, aiding the understanding of infection mechanisms.
Stukalov A et al., Nature, 2021
Sample Submission Suggestions
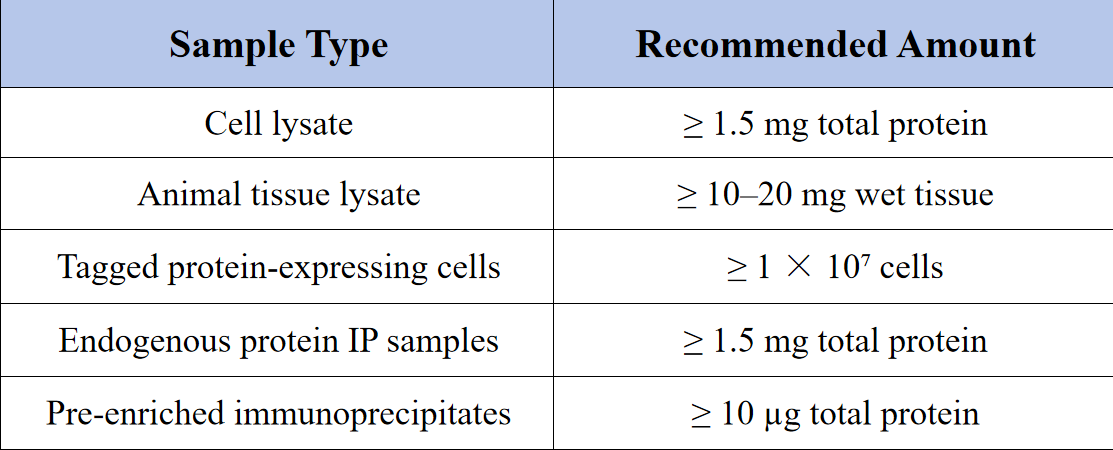
Notes: To ensure data quality, please avoid using components that interfere with mass spectrometry analysis, such as SDS, detergents, or high concentrations of glycerol. Samples should be stored at −80°C and shipped on dry ice.
If your samples involve alternative sources or non-standard preparation protocols, please contact us in advance. We will provide tailored pre-processing guidance based on your specific project needs.
Deliverables
What Could be Included in the Report?
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Related Services
SILAC Based Co-IP-MS for Protein Interaction Analysis Service
MS-Based Protein-Protein Interaction Analysis Service
How to order?

