





LC-MS Based Intact Protein Analysis Service
Intact MS (Intact protein mass spectrometry) provides a holistic profile of a biotherapeutic, helping to confirm predictive masses, analyze product heterogeneity, and identify impurities. In recent years, liquid chromatography mass spectrometry (LC-MS) has become an indispensable tool for characterization and monitor of biotherapeutics at all stages from their development to commercialization. Capabilities such as discovery screening, process development, and formulation/stability evaluation enable high-volume sample generation, and analytical solutions that enable high-throughput Intact MS analysis, while impurity analysis can help match these requirements.
Intact MS is uniquely positioned in areas where in vivo primary amino acid sequence integrity needs to be measured or to monitor cleavage forms/biotransformation products. This is especially important for complex biotherapeutics, which are more likely to put into in vivo quality stability test. Intact MS helps to provide more comprehensive information on physicochemical properties of drugs and gain insight into the structure of their targets. Intact MS also specifically address the shortcomings of the digestion method. First, it is no longer necessary to select characteristic peptides. Second, due to the elimination of the digestion step, the variability of the Intact MS assay may be reduced compared to enzymatic digestion-based assays. From a stability standpoint, peptide assays don't really answer many questions about the long-term stability of molecules in vivo, whereas Intact MS assays can observe whether the Intact MS is stable or not, and may even help identify sites of instability. The combination of Intact MS and traditional antibody subunit monitoring provides a 100% sequence coverage, which is not possible with other monitoring methods.
Mass spectrometry (MS) of proteins can be divided into three types, which are bottom-up method, middle-down method, and top-down method. Enzymes such as trypsin are used to digest intact proteins into peptides in the Bottom-up MS method. For antibody subunit analysis, a middle-down strategy can be employed, i.e. disulfide reduction and/or partial digestion, which is useful for prime variants in subunit localization, such as antibody-drug conjugates (ADCs) or fusion proteins. Top-down MS can directly obtain the information of intact protein mass and post-translational modifications (PTMs) based on LC-MS.
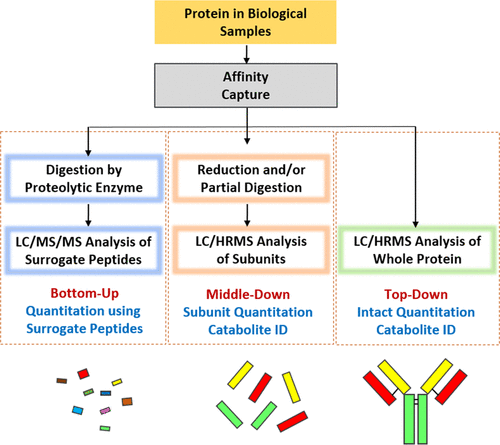
Figure 1. Protein MS Strategy [3]
Top-down MS technology mainly includes three aspects: intact protein separation, MS detection and bioinformatics technology. Protein separation is mainly to reduce the complexity of MS analysis and injection, and improve the signal-to-noise ratio (SNR) of MS data. Efficient separation technology is the prerequisite for large-scale protein variant identification, and effective protein separation methods are mainly divided into off-line and on-line methods, the former is represented by the "four-dimensional offline separation" method, and the latter is weak cation exchange (WCX) combined with hydrophilic interaction liquid chromatography (HILIC) on-line separation method as representatives. Effective "on-line" (i.e., directly coupled to MS) separation is important for the analysis of complex samples. In the case of liquid chromatography (LC), a range of separation principles can be used. For example, analytes can be separated based on their size (size-exclusion chromatography, SEC), charge under different salt or pH conditions (ion-exchange chromatography, IEX), hydrophobic interaction with the column material (reversed-phase liquid chromatography, RPLC), hydrophilic interaction (HILIC), or based on affinity for a particular column material (affinity chromatography, AC). MS analysis mainly includes two aspects: measuring the accurate molecular mass of intact proteins and obtaining fragmentation spectra of proteins, the most critical of which MS fragmentation is the core of reliable identification scoring algorithms. Bioinformatics analysis must rely on efficient algorithms and software, and due to the complexity of top-down protein MS, algorithms and software tools face great challenges in addition to database searches, especially the distinction between signal and noise, and charge state deconvolution.
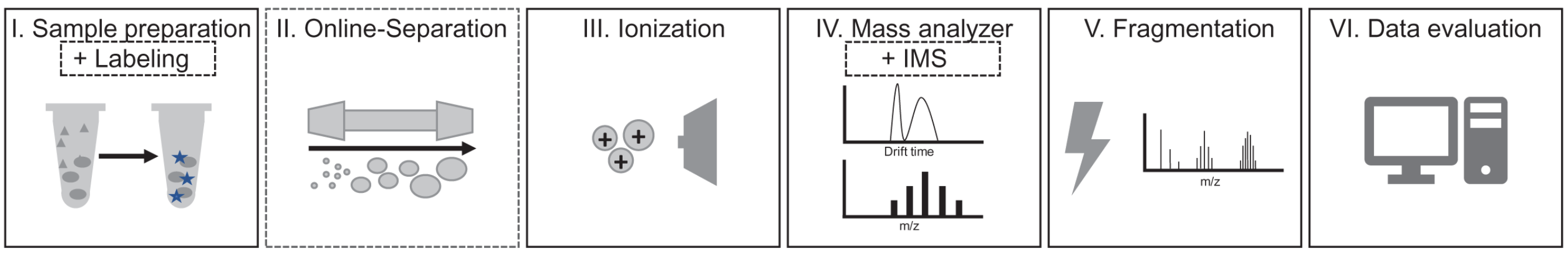
Figure 2. Main Steps Required for Top-Down Protein MS Experiment
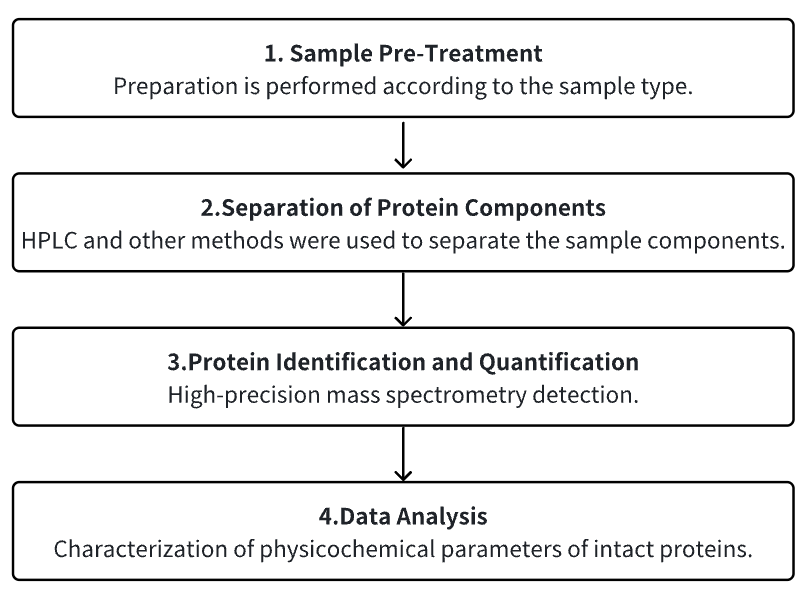
Analysis Workflow
1. Sample Pre-Treatment
2. Fractionation of Protein Components
3. MS Detection
4. Data Analysis
Service Advantages
1. Identification/Quantification/Modification Identification of Intact Proteins from Multiple Types of Samples
2. High Confidence, High-Precision MS Detection
3. Comprehensive Bioinformatics Analysis
Sample Results
1. MS Detection of Non-Covalent Protein-Drug Complexes in Tissues from Orally Administered Rats [5]
Native ambient MS was used to detect the intact protein-drug complex formed in vivo between endogenous liver fatty acid-binding protein (FABP1) and orally dosed bezafibrate can be detected directly from ex vivo liver tissue. The spatial distribution of protein-drug complexes is visualized by native nanospray desorption electrospray ionization (nano-DESI).
Figure 3. Native LESA HCD MS/MS Spectra of the [FABP1+ Bezafibrate] Ion
2. High-Throughput Intact Protein Analysis for Drug Discovery Using Infrared Matrix-Assisted Laser Desorption Electrospray Ionization Mass Spectrometry MS (IR-MALDESI-MS) [6]
MS is the primary analytical tool used to characterize proteins in the biopharmaceutical industry. Electrospray ionization (ESI) combined with liquid chromatography (LC) is the current gold standard for intact protein analysis. However, the inherent speed limitations of LC-MS make it impossible to analyze a large sample numbers (>1000) in a day . IR-MALDESI-MS is an ambient ionization MS technique that has recently been established as a platform for high-throughput small molecule analysis. The application of this system to the analysis of intact proteins typically performed in the drug discovery process is reported in the study. Proteins with a wide molecular weight range of 10-150 kDa were detected on the system, which is more tolerant to salts and buffers compared to ESI.
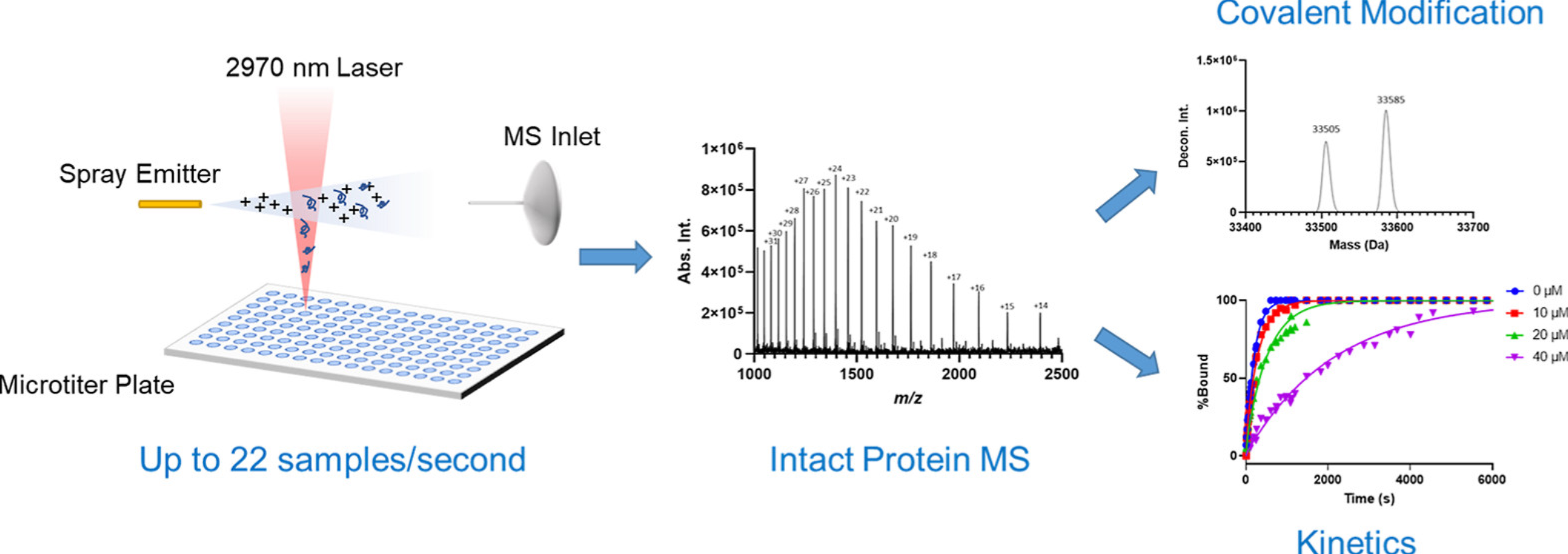
Figure 4. IR-MALDESI-MS Analysis of Intact Proteins
3. Evaluation of Complete Protein Profiling for Quantitative Analysis of Protein Therapy [7]
The complexity of the protein therapeutics manufacturing process and the sensitivity of these drugs to storage conditions can lead to the presence of byproducts, such as including protein variants cuased by truncations, degradation, and differentially modified that are difficult to be detected by peptide analysis. Intact MS can be used to identify intact protein composition, including PTMs. Still, it can also generate chemical fingerprints of different protein present in a given sample. In this work, the effects of multiple charge states, multiple isotopes for each charge state, and operational resolution on the applicability of intact MS for quantitative analysis using insulin and growth hormone as model systems were systematically evaluated. Standard curves can be generated using absolute intensity data or by using the relative ratio between analytes and internal standards. These methods demonstrate the effectiveness of quantitative of intact MS for the analysis of protein therapeutics, thus providing a basis for future comparative methods.
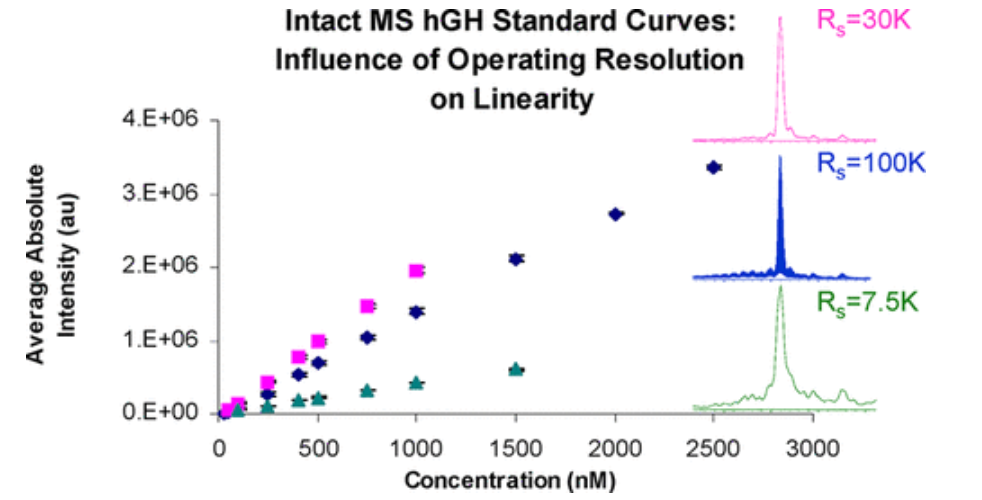
Figure 5. Intact MS-Based Quantification of Macromolecular Proteins (Insulin and Growth Hormone)
4. High-Resolution MS to Characterize the in vivo Biotransformation of Intact ADCs [8]
ADCs represent a promising class of therapies for targeted delivery of highly potent cytotoxic drugs to tumor cells to enhance biological activity while minimizing their side effects. ADCs are composed of small molecules and large molecules, so they have a complex molecular structure. In vivo biotransformation can further increase the complexity of ADCs, which presents a great challenge for bioanalytical analysis. Quadrupole time-of-flight MS (Q-TOF MS) with ESI has been widely used to characterize intact ADCs. However, the interpretation of ADC biotransformations with small changes in intact molecular mass remains a limitation due to the insufficient mass resolution and accuracy of Q-TOF MS. Therefore, the study investigated the in vivo biotransformation of multi-site-specific THIOMAB™ drug conjugates (TDCs) in intact forms using a high-resolution, accurate-mass (HR/AM)MS approach. Compared to traditional Q-TOF MS, HR/AM Orbitrap MS provides a more comprehensive identification of ADC biotransformations. It is particularly beneficial for characterizing ADC modifications with small mass changes, such as partial drug loss and hydrolysis.
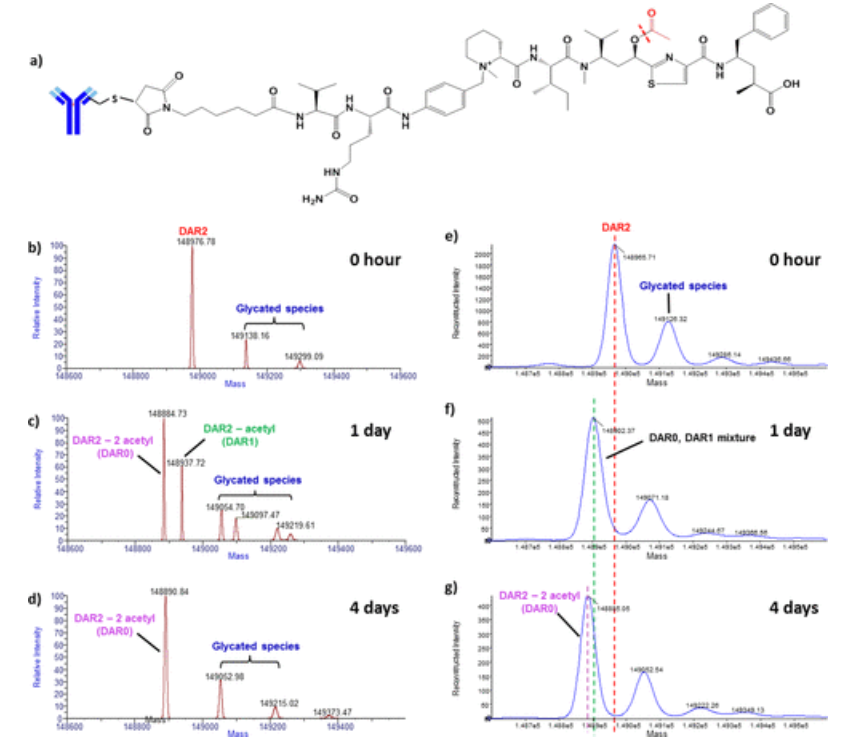
Figure 6. Characterization of Anti-CD22 MC-vc-PAB Tubulysin M TDC1 Deacetylation
Sample Submission Requirements
1. Provide pre-purified samples whenever possible.
2. Try to avoid impurity pollution.
Services at MtoZ Biolabs
1. Complete Experimental Protocol
2. Relevant Instrument Parameters
3. Raw MS Data
4. Detailed Information Report on the Identification of Protein Macromolecules (including: accurate molecular weight determination, primary sequence confirmation, PTMs, etc.)
Applications
1. Intact MS is used to quantify ADCs at intact protein levels in biological samples.
Preclinical and in vitro characterization of ADCs involves the development of several bioanalytical methods to address many drug exposure issues. Current pharmaceutical industry methods require at least three different assays to be run, i.e., total mAb, conjugated or conjugated mAb, and free payload assays. In addition to quantification, the Intact MS method can provide useful information about the drug-to-antibody ratio (DAR) distribution, average DAR, and mAb partial primary sequence modifications.
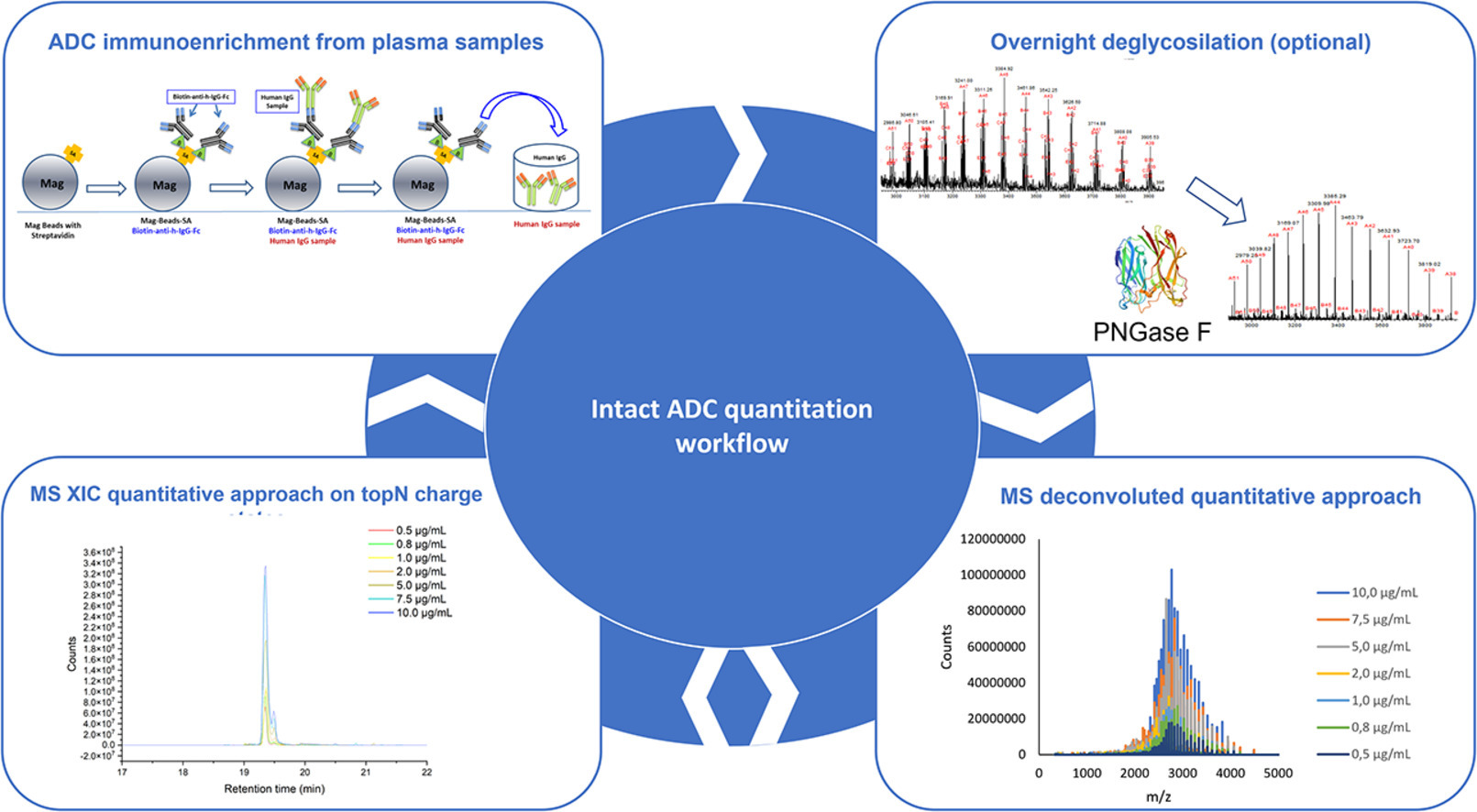
Figure 7. Intact MS Workflow for ADCs Detection [9]
2. Application of intact MS in preclinical pharmacokinetic studies. [10]
A new workflow has been proposed by hybridizing the ligand binding assay (LBA) with liquid chromatography-high-resolution MS (LC-HRMS) to quantify hIgG1 directly at the intact protein level. The assay was validated in terms of selectivity, sensitivity, accuracy/precision, carryover, dilution linearity, and reproducibility.
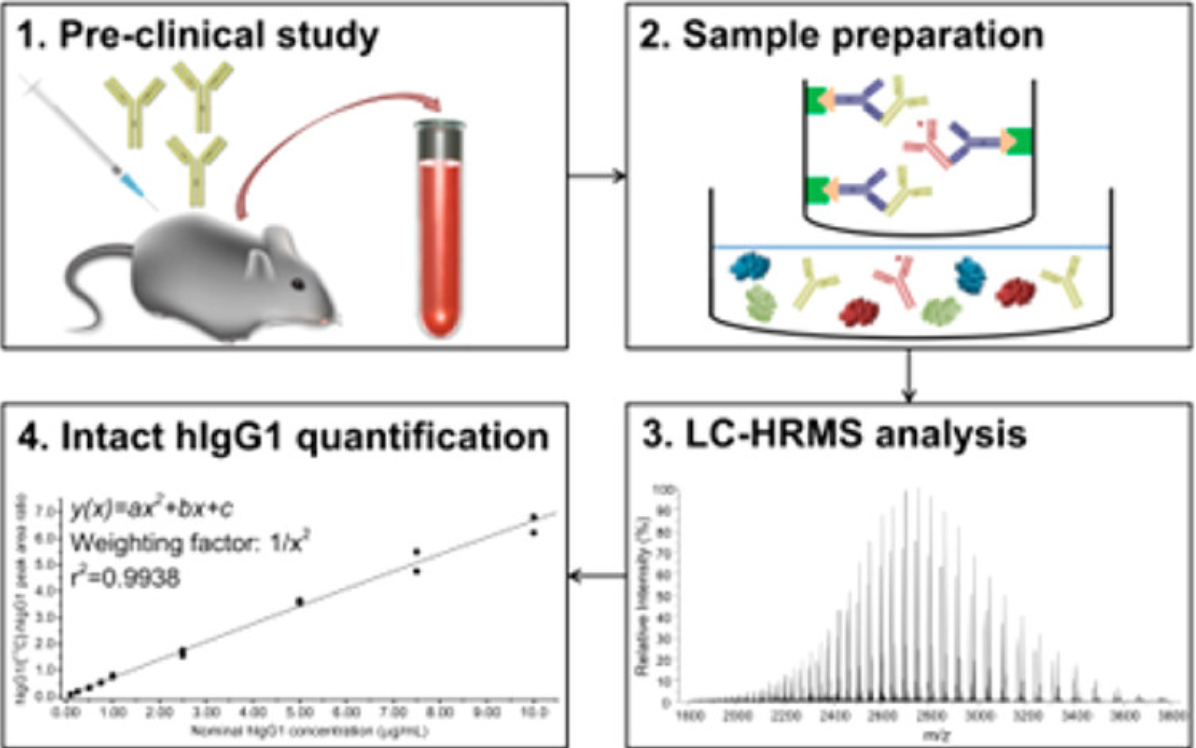
Figure 8. Intact Protein MS was Used to Analyze Changes in Human Immunoglobulin G1 Levels in Serum
3. Intact MS for high-resolution glycoform analysis of intact therapeutic proteins.
Glycosylation is considered a critical quality attribute of therapeutic proteins. Protein heterogeneity introduced by glycosylation includes differences in the nature, number, and location of glycans. The analysis of glycans and glycopeptides provides information on glycan composition and/or location, while intact glycoprotein analysis allows for the assignment of individual protein types and cooccurring modifications.
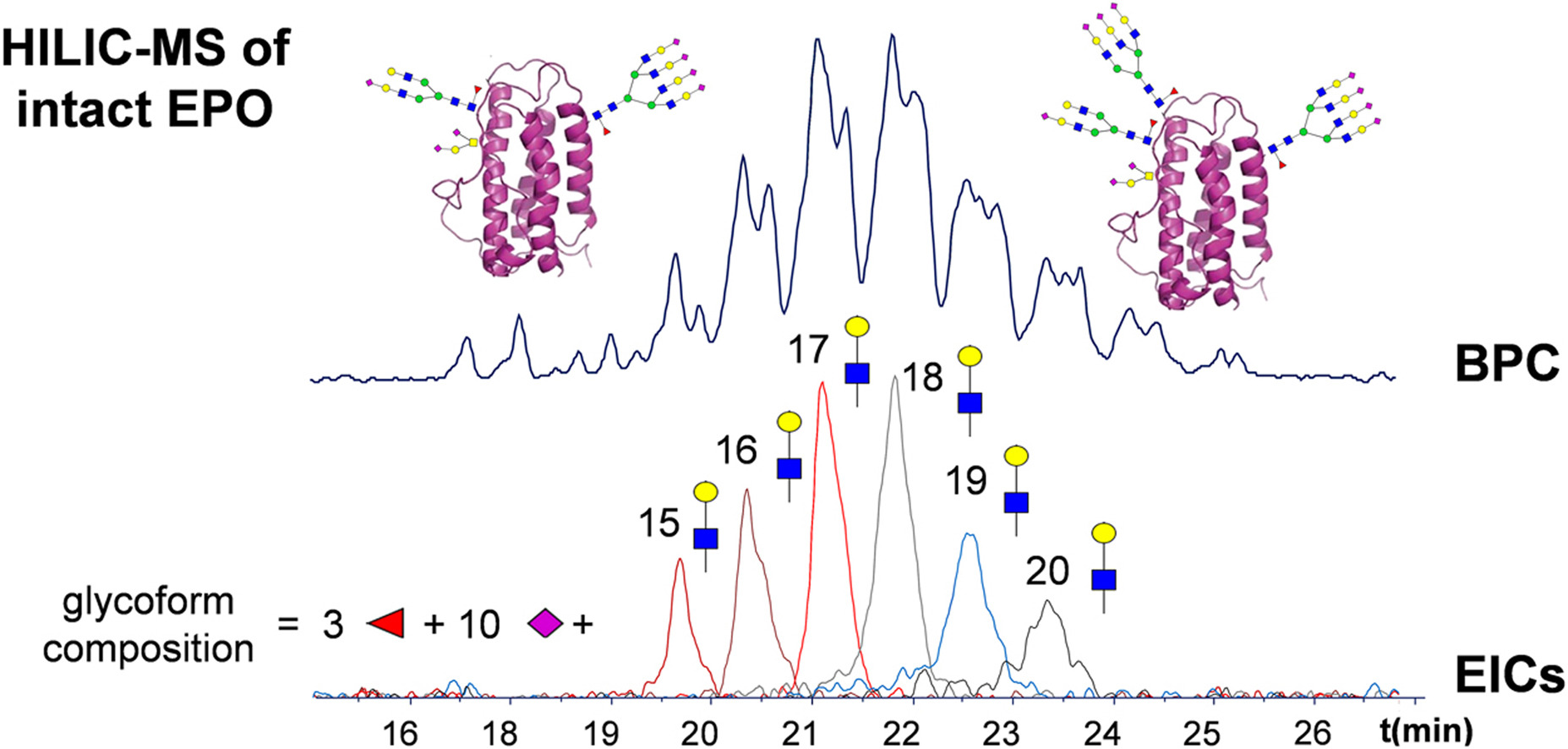
Figure 9. Intact Protein MS Provides Drug Glycoform Profile [11]
FAQ
Q1: What should I pay attention to when preparing protein samples for Intact MS analysis?
Non-volatile buffers and high salt concentrations. These conditions are not MS-friendly, especially with traditional ESI, as they can induce ion suppression and/or form non-specific adducts, which necessitates desalting or upstream separations of protein solutions to obtain high-quality data. On the other hand, it is also important to keep the protein in as natural conditions as possible to preserve the non-covalent interactions in protein-protein or protein-ligand complexes. Ammonium acetate aqueous solution (AmAc) is the solvent of choice because it has a pH of about 7, ionic strength comparable to NaCl, and volatility under ESI conditions. It also provides protons for the electrospray process, promoting the production of [M+nH]n+ ions rather than adducts with Na+ or other non-volatile substances.
References
[1] Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci. 2019 Aug;40(8):592-604. doi: 10.1016/j.tips.2019.06.004. Epub 2019 Jul 15. Erratum in: Trends Pharmacol Sci. 2019 Oct;40(10):801. PMID: 31320117.
[2] Pinzi L, Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int J Mol Sci. 2019 Sep 4;20(18):4331. doi: 10.3390/ijms20184331. PMID: 31487867; PMCID: PMC6769923.
[3] Kellie JF, Tran JC, Jian W, Jones B, Mehl JT, Ge Y, Henion J, Bateman KP. Intact Protein Mass Spectrometry for Therapeutic Protein Quantitation, Pharmacokinetics, and Biotransformation in Preclinical and Clinical Studies: An Industry Perspective. J Am Soc Mass Spectrom. 2021 Aug 4;32(8):1886-1900. doi: 10.1021/jasms.0c00270. Epub 2020 Sep 15. PMID: 32869982.
[4] Habeck T, Lermyte F. Seeing the complete picture: proteins in top-down mass spectrometry. Essays Biochem. 2023 Mar 29;67(2):283-300. doi: 10.1042/EBC20220098. PMID: 36468679.
[5] Illes-Toth E, Hale OJ, Hughes JW, Strittmatter N, Rose J, Clayton B, Sargeant R, Jones S, Dannhorn A, Goodwin RJA, Cooper HJ. Mass Spectrometry Detection and Imaging of a Non-Covalent Protein-Drug Complex in Tissue from Orally Dosed Rats. Angew Chem Int Ed Engl. 2022 Sep 5;61(36):e202202075. doi: 10.1002/anie.202202075. Epub 2022 Jul 13. PMID: 35830332; PMCID: PMC9542108.
[6] Pu F, Ugrin SA, Radosevich AJ, Chang-Yen D, Sawicki JW, Talaty NN, Elsen NL, Williams JD. High-Throughput Intact Protein Analysis for Drug Discovery Using Infrared Matrix-Assisted Laser Desorption Electrospray Ionization Mass Spectrometry. Anal Chem. 2022 Oct 4;94(39):13566-13574. doi: 10.1021/acs.analchem.2c03211. Epub 2022 Sep 21. PMID: 36129783.
[7] Gucinski AC, Boyne MT 2nd. Evaluation of intact mass spectrometry for the quantitative analysis of protein therapeutics. Anal Chem. 2012 Sep 18;84(18):8045-51. doi: 10.1021/ac301949j. Epub 2012 Sep 4. PMID: 22916992.
[8] He J, Su D, Ng C, Liu L, Yu SF, Pillow TH, Del Rosario G, Darwish M, Lee BC, Ohri R, Zhou H, Wang X, Lu J, Kaur S, Xu K. High-Resolution Accurate-Mass Mass Spectrometry Enabling In-Depth Characterization of in Vivo Biotransformations for Intact Antibody-Drug Conjugates. Anal Chem. 2017 May 16;89(10):5476-5483. doi: 10.1021/acs.analchem.7b00408. Epub 2017 May 3. PMID: 28429938.
[9] Barbero LM, Ianni AD, Molinaro F, Cowan KJ, Sirtori FR. Hybrid liquid chromatography high resolution and accuracy mass spectrometry approach for quantification of antibody-drug conjugates at the intact protein level in biological samples. Eur J Pharm Sci. 2023 Sep 1;188:106502. doi: 10.1016/j.ejps.2023.106502. Epub 2023 Jun 17. PMID: 37336420.
[10] Lanshoeft C, Cianférani S, Heudi O. Generic Hybrid Ligand Binding Assay Liquid Chromatography High-Resolution Mass Spectrometry-Based Workflow for Multiplexed Human Immunoglobulin G1 Quantification at the Intact Protein Level: Application to Preclinical Pharmacokinetic Studies. Anal Chem. 2017 Feb 21;89(4):2628-2635. doi: 10.1021/acs.analchem.6b04997. Epub 2017 Feb 9. PMID: 28192936.
[11] Domínguez-Vega E, Tengattini S, Peintner C, van Angeren J, Temporini C, Haselberg R, Massolini G, Somsen GW. High-resolution glycoform profiling of intact therapeutic proteins by hydrophilic interaction chromatography-mass spectrometry. Talanta. 2018 Jul 1;184:375-381. doi: 10.1016/j.talanta.2018.03.015. Epub 2018 Mar 8. PMID: 29674057.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Intact Protein Analysis Service
How to order?

