





Global Proteomic Quantification Service
- Labeling Quantification (TMT/iTRAQ)
- Label-Free Quantification (LFQ/DIA)
Global Proteomic Quantification Service performs comprehensive quantitative analysis of all proteins in biological samples using mass spectrometry technology. It can simultaneously identify and quantify thousands of proteins, offering insights into protein expression, regulation, and function. Unlike traditional quantitative methods, Global Proteomic Quantification delivers in-depth analysis of the entire proteome without the need to preselect or focus on specific target proteins, thus providing a holistic, system-wide data perspective. Utilizing mass spectrometry technology, MtoZ Biolabs offers Global Proteomic Quantification Service for a wide range of biological samples, enabling accurate and reliable protein quantification to help researchers gain deeper understanding of protein roles in various biological processes.
Technical Principles
Global Proteomic Quantification Service utilizes high-resolution mass spectrometry in combination with data-dependent acquisition (DDA) and data-independent acquisition (DIA) modes to perform comprehensive quantitative analysis of the entire proteome. Through precise peptide and protein spectrum comparison and quantification, combined with highly sensitive quantitative analysis methods, the quantitative accuracy of proteins in each sample can be ensured. Accurate quantitative results can be obtained by using labeling methods (such as TMT, iTRAQ, etc.) and non-labeling methods.
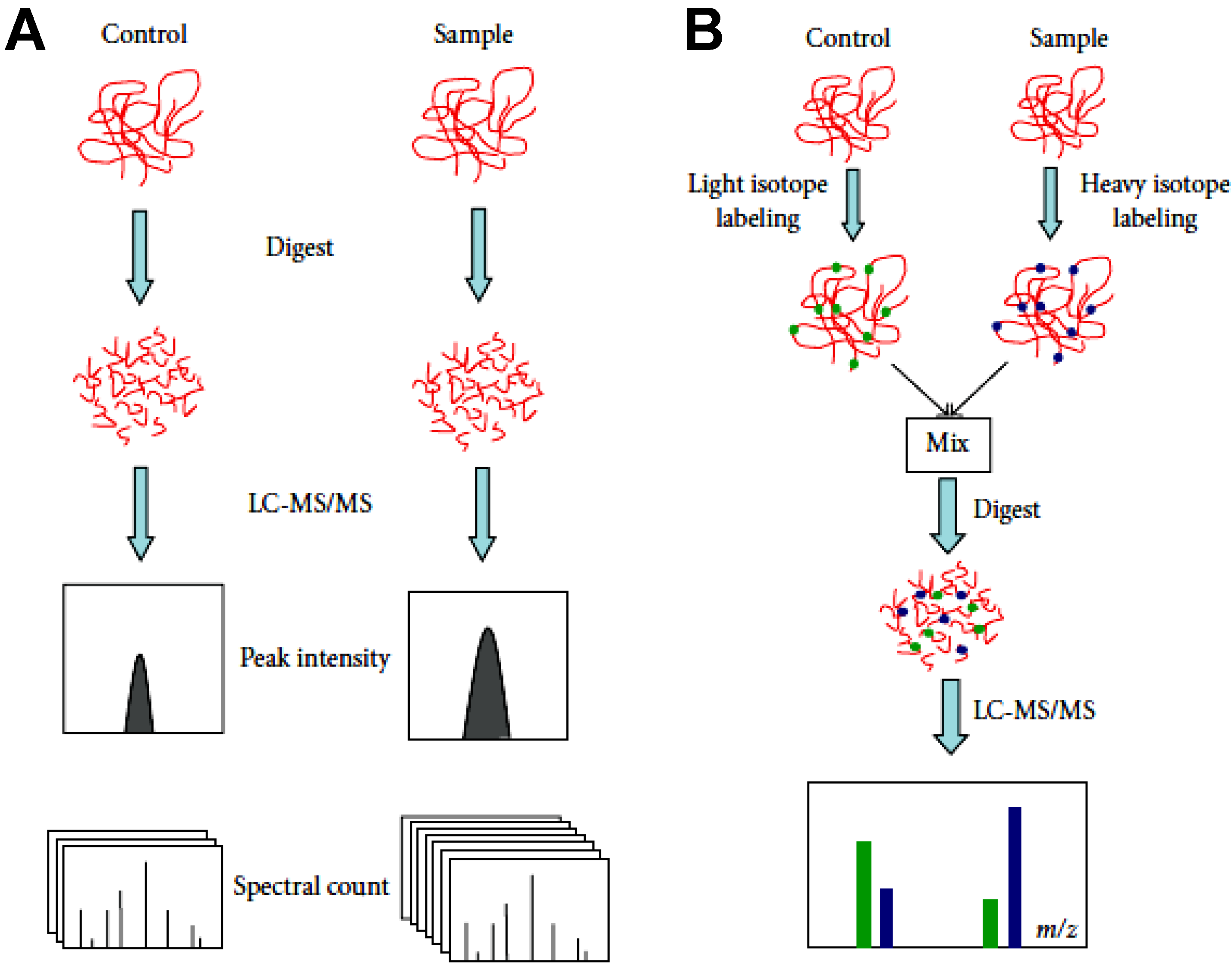
Deracinois B. et al. Proteomes. 2013.
Figure 1. Comparison of label-free and labeling quantification
Analysis Workflow
1. Sample Preparation
Processing and protein extraction are performed according to sample type.
2. Digestion and Labeling
Proteins are digested into peptides, and labeling techniques can be used for quantification.
3. Mass Spectrometry Analysis
Proteomic analysis is performed using LC-MS/MS, with data acquired in either DDA or DIA mode.
4. Data Analysis
Specialized analytical software is used to process the data and quantify proteins, including database search and quantitative result interpretation.
5. Reporting
A comprehensive quantitative analysis report is provided, detailing relative abundances of proteins and peptides, standard deviations, confidence intervals, and other key metrics.
Service Advantages
1. Advanced Analysis Platform: MtoZ Biolabs established an advanced Global Proteomic Quantification Service platform, guaranteeing reliable, fast, and highly accurate analysis service.
2. One-Time-Charge: Our pricing is transparent, no hidden fees or additional costs.
3. High-Data-Quality: Deep data coverage with strict data quality control. AI-powered bioinformatics platform integrates all Global Proteomic Quantification Service data, providing clients with a comprehensive data report.
4. Labeling and Label-Free Quantification Methods: Flexible labeling and label-free quantification services are provided based on the specific research requirements.
FAQ
Q. How to choose the appropriate quantification technique (Labeling vs. Label-Free Quantification) in Global Proteomic Quantification studies?
Quantitative proteomics commonly employs two strategies: Labeling quantification (e.g., TMT, iTRAQ) and label-free quantification (Label-Free Quantification, LFQ).
This method is suitable for relative quantification analysis across multiple samples and allows for direct comparison of several samples in a single experiment. It has the advantages of high throughput and reduced batch effects, but the cost is relatively high, and there may be ion suppression effects on the detection of low-abundance proteins.
Ideal for large-scale proteome quantification analysis, this method is not limited by a fixed number of samples and can detect proteins across a broad dynamic range. It is well-suited for exploratory research or low-abundance protein analysis, but it requires higher sample reproducibility to ensure accuracy.
Q. How to ensure high-quality protein samples during protein extraction and sample processing?
The quality of protein extraction and processing directly impacts the accuracy and reproducibility of quantification. Key considerations include:
1. Sample Processing
Use low-temperature lysis to minimize protein degradation.
Add protease inhibitors to prevent protein hydrolysis.
Select an appropriate lysis method for the sample type, such as SDS lysis for tissue samples and high-salt or detergent-based methods for plasma samples.
2. Protein Quantification
Use BCA or Bradford method for quantification to ensure uniform protein concentration in all samples to avoid quantitative analysis affected by concentration differences. And sufficient sample volume is required.
3. Protein Digestion
Perform trypsin digestion, controlling the digestion time (typically 8-16 hours) to achieve uniform peptide fragments.
Use C18 columns for desalting to remove contaminants and improve mass spectrometry detection sensitivity.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
Case Study
This study used Global Proteomic Quantification to systematically screen and identify protein degradation targets regulated by the mTORC1-CTLH E3 ligase complex (E3 ligase). The results indicate that HMG-CoA synthase 1 (HMGCS1) is regulated by the CTLH E3 ligase via the Pro/N-degron pathway and is influenced by the mTORC1 signaling pathway. This study revealed the protein homeostasis regulation mechanism of mTORC1 on HMGCS1, a key enzyme in cholesterol synthesis, providing important clues for exploring new functions of mTORC1 in metabolic regulation, and may provide new therapeutic strategies for the research of metabolic-related diseases and cancer.
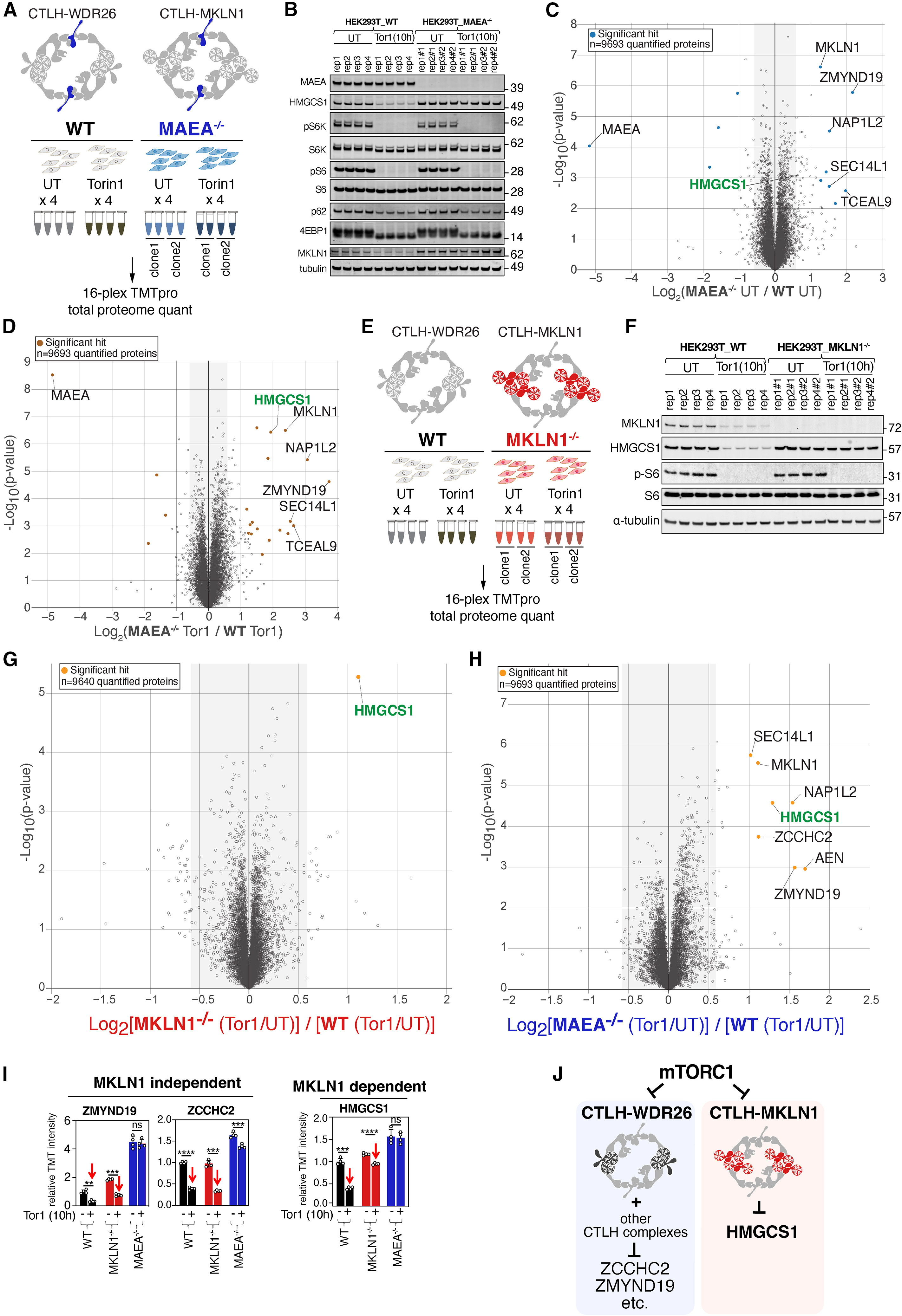
Yi S A. et al. Molecular Cell. 2024.
Figure 2. Global proteomic analysis to study the CTLH E3-dependent proteome changes during mTOR inhibition
How to order?

