





From TMT to SWATH: Evolution of Quantitative Proteomics Technologies
-
Label-based approaches: These methods employ chemical reagents to tag peptides from distinct samples. Representative techniques include TMT and iTRAQ, offering advantages such as high throughput and quantitative precision.
-
Label-free approaches: These techniques quantify peptides across samples based on signal intensity or spectral counting, as exemplified by LFQ and SWATH. They are particularly suitable for large-scale studies with cost constraints.
-
Each sample is labeled with a distinct TMT reagent;
-
Labeled samples are pooled and analyzed simultaneously via mass spectrometry;
-
During MS2 acquisition, reporter ions are released and measured to achieve quantification.
-
High throughput: Enables up to 18-plex multiplexed analysis;
-
High consistency: Co-analysis of all samples minimizes inter-batch variability;
-
Suited for complex biological matrices: Including clinical specimens and heterogeneous tumor tissues.
-
Employ high-resolution mass spectrometers to enhance peptide identification selectivity;
-
Integrate advanced acquisition methods such as SPS-MS3 to improve quantification specificity;
-
Refine sample preparation protocols, including peptide fractionation and purification, to reduce background complexity and enhance quantification accuracy.
-
Comprehensive coverage without data loss: mitigates the omission of relevant signals due to preferential analysis of only the most intense precursors;
-
High reproducibility: employs a consistent acquisition strategy, making it suitable for large-scale clinical cohort studies;
-
Reusability of data: complete spectral information is retained, facilitating retrospective analysis and multi-omics data integration.
-
Construct high-quality, species-specific reference spectral libraries to enhance the efficiency of peptide identification and matching;
-
Apply advanced data processing platforms (e.g., Spectronaut, DIA-NN) to improve feature extraction and peak detection accuracy;
-
Ensure batch-to-batch consistency during sample preparation to minimize systematic variability introduced during sample handling.
In proteomics research, quantitative analysis of proteins plays a pivotal role in elucidating cellular states, understanding disease mechanisms, and identifying potential biomarkers. With the rapid advancement of mass spectrometry (MS) technologies, protein quantification approaches have evolved significantly—from early 2D gel-based techniques to modern high-throughput platforms such as Tandem Mass Tag (TMT) and Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH). This review outlines the developmental trajectory of quantitative proteomics technologies, with a focus on the principles, advantages, and appropriate application contexts of TMT and SWATH methodologies.
Two Principal Approaches to Protein Quantification
Current mass spectrometry-based quantitative strategies can be broadly classified into two categories:
The selection of an appropriate quantification strategy depends on factors such as experimental design, sample throughput, budget considerations, and required data accuracy.
TMT: A Mainstream Technology for High-Precision Multiplexed Quantification
TMT is an isobaric labeling strategy widely employed to assess protein expression differences across multiple samples.
1. Principle
2. Advantages
3. Technical Challenges and Optimization Strategies
Despite its strengths in throughput and precision, TMT-based quantification faces notable challenges, the most prominent being quantification ratio compression. This artifact primarily arises from co-isolated peptide interference during the MS2 stage, leading to underestimated inter-sample differences.
To mitigate these effects, several optimization strategies are commonly applied:
SWATH: Comprehensive and Unbiased Scanning—Heralding a New Era of Data-Independent Acquisition
SWATH represents a protein quantification approach based on the Data-Independent Acquisition (DIA) strategy. In contrast to the Data-Dependent Acquisition (DDA) mode commonly employed in TMT workflows, SWATH does not preselect precursor ions during data collection. Instead, the m/z range is segmented into multiple consecutive windows, allowing for the unbiased acquisition of all fragment ion spectra.
1. Technical Advantages
2. Technical Challenges and Optimization
Although SWATH, as a representative DIA method, offers advantages such as unbiased full-spectrum acquisition and retrospective data analysis, it poses significant implementation challenges due to the complexity of its data structure and the high computational demands for accurate interpretation.
Optimization Strategies:
Comparative Overview of TMT and SWATH Technologies
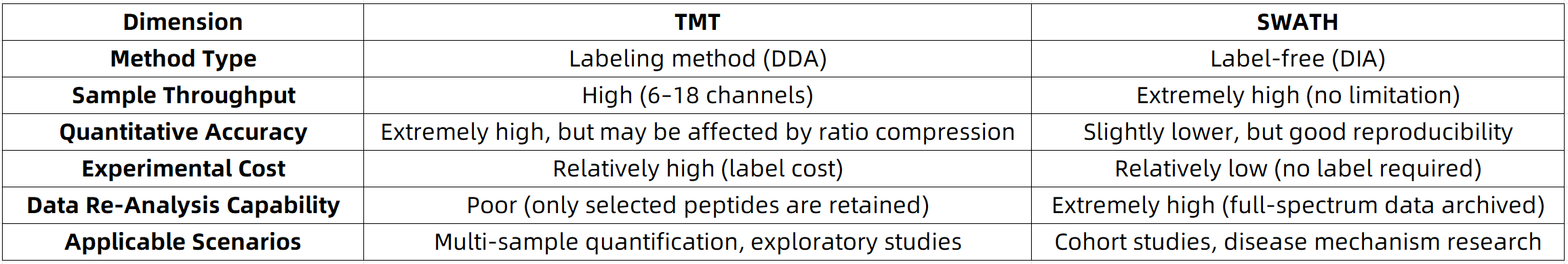
As multi-omics integration and artificial intelligence–assisted analysis continue to advance, protein quantification technologies are evolving toward enhanced sensitivity, expanded dynamic range, and broader applicability. TMT and SWATH, as two prevailing quantification strategies, each offer unique strengths and are complementary in practice. MtoZ Biolabs remains committed to supporting the advancement of life science research by providing expert technical solutions for deeper molecular-level analysis. For detailed information on our SWATH-based quantitative proteomics services and pricing, please feel free to contact us.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

