





Deamidation Proteomics Service
Deamidation Proteomics Service refers to a proteomics-level service that utilizes high-resolution mass spectrometry and other technological approaches to systematically identify and quantify deamidation sites in proteins. This service can not only be used to reveal structural changes and degradation pathways of proteins, but also be applied in areas such as biopharmaceutical quality control, aging-related disease research, and investigations into the mechanisms of neurodegenerative diseases.
Protein deamidation is a common post-translational modification (PTM) in which asparagine (Asn) and glutamine (Gln) residues undergo spontaneous, non-enzymatic conversion to acidic amino acids (aspartic acid or glutamic acid). This modification plays a significant regulatory role in various biological processes. Deamidation can profoundly impact a protein’s charge state, spatial conformation, stability, and even its biological function. It is increasingly recognized in studies on protein aging, degradation pathways, drug stability, and neurodegenerative diseases.
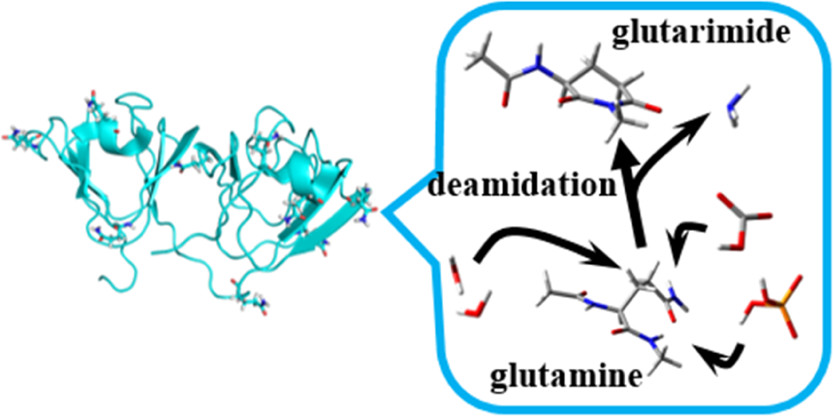
Kato K. et al. ACS Omega. 2019.
Deamidation reactions increase the molecular weight of proteins by approximately 0.984 Da, resulting in a decrease in isoelectric point (pI), alterations in charge state, and even conformational rearrangements. In mass spectrometry analysis, this subtle yet detectable mass shift provides a theoretical basis for the precise localization of modification sites.
Based on advanced chromatography and mass spectrometry platforms, MtoZ Biolabs offers Deamidation Proteomics Service to help clients comprehensively identify and quantify deamidation modification sites in proteins, enabling in-depth analysis of their impact on protein function and stability.
Analysis Workflow
The main workflow of Deamidation Proteomics Service is as follows:
1. Sample Preparation
Buffer exchange and concentration adjustment are performed based on sample characteristics. Proteolytic digestion is then carried out using proteases.
2. Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis
Peptides are separated and analyzed using a nano-LC system coupled with high-resolution mass spectrometry to identify deamidation modification sites.
3. Data Processing
Specialized software platforms are used for database searching and modification site identification. Functional annotation and pathway analysis are performed using bioinformatics tools.
4. Result Reporting
A detailed analysis report is provided, including identification results of deamidation sites, quantitative analysis data, functional annotation information, and related charts.
Applications
Deamidation Proteomics Service offers valuable applications in multiple areas:
Biopharmaceutical Quality Control
Detection of deamidation modifications in antibody-based drugs and recombinant proteins to assess drug stability, heterogeneity, and efficacy.
Protein Aging and Stability Studies
Investigation of the natural aging mechanisms and degradation pathways of proteins.
Disease Research
Exploration of the role of deamidation in neurodegenerative diseases, cancer, and other conditions to identify potential biomarkers.
Protein Function Studies
Analysis of the impact of deamidation on protein structure and function, uncovering its biological significance in cellular signaling, metabolic regulation, and other processes.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Total Ion Chromatogram & Quality Control Assessment (project-dependent)
4. Data Analysis, Preprocessing, and Estimation (project-dependent)
5. Bioinformatics Analysis
6. Raw Data Files
FAQ
Q. How Can You Ensure that the Detected Deamidation is a True Biological Event Rather Than a Sample Preparation-Induced Artifact?
We place high importance on the biological authenticity of deamidation data. During sample preparation, we use low-pH buffers, low-temperature rapid digestion, and anti-deamidation buffer systems to effectively suppress nonspecific in vitro deamidation. We also recommend that clients include non-digested or briefly processed control groups. By integrating chromatographic retention time, peak shape characteristics, and replicate comparisons, we can assess whether deamidation originates from in vivo processes, thereby ensuring that the results reflect true physiological relevance.
Q. Is it Possible to Accurately Quantify Deamidation and Compare Modification Levels Across Different Samples?
Yes. MtoZ Biolabs supports multiple quantification strategies, including label-free quantification (LFQ) and labeling-based quantification (e.g., TMT/iTRAQ), which can be flexibly adapted to various experimental designs. Using high-resolution mass spectrometry coupled with high-performance liquid chromatography, along with specialized data analysis platforms such as MaxQuant and Proteome Discoverer, we can precisely extract peak areas of deamidated peptides and perform statistical analysis. This enables reliable comparisons of deamidation levels across different sample groups or treatment conditions and facilitates interpretation of the relationship between modifications and functional or pathological states.
Case Study
This study employed high-resolution mass spectrometry to perform a systematic post-translational modification (PTM) analysis of amyloid plaques and surrounding regions in the brains of Alzheimer’s disease transgenic mice. Special focus was given to various PTMs, including deamidation. The results revealed that deamidation was one of the three most enriched modifications within the plaques, suggesting a strong association with protein aggregation, stability alterations, and disease progression. These findings indicate that deamidation may play a key role in Alzheimer’s disease pathology and provide valuable insights for the mechanistic study of neurodegenerative disorders.
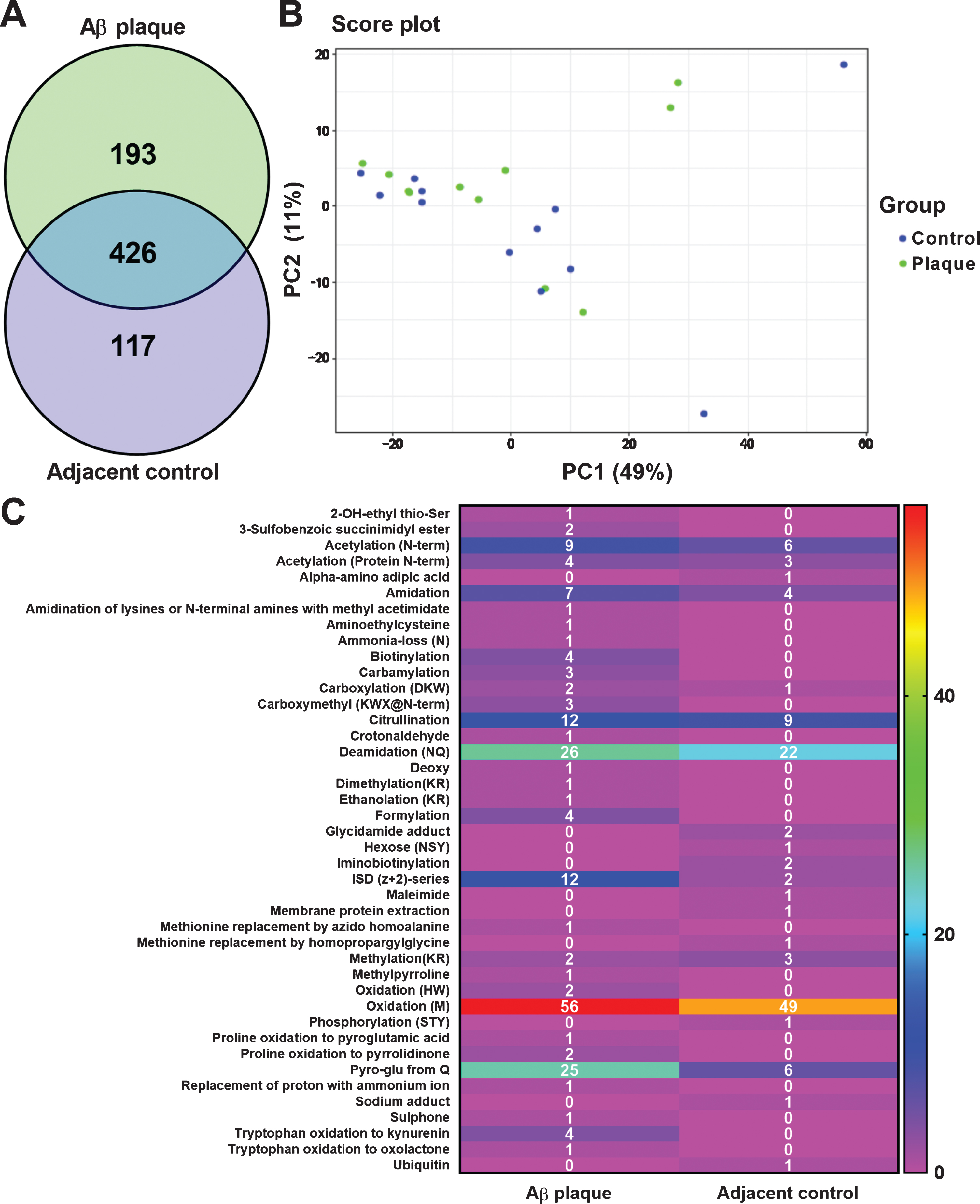
Bastrup J. et al. Journal of Alzheimer's Disease. 2020.
How to order?

