





De Novo Sequencing vs Database-Assisted MS: Choosing the Right Protein Sequence Method
-
Reference requirement: Does the method need a known sequence?
-
Project goal: Is the goal discovery, confirmation, or documentation?
-
Interpretation burden: How much expert review is required?
-
Turnaround and cost profile: How do sample complexity and reporting needs affect timeline and budget?
-
no reliable reference sequence exists
-
database search fails despite acceptable spectral quality
-
the sample is a proprietary, novel, or otherwise unannotated protein
-
sequence recovery must come from purified protein material alone
-
the sample comes from a well-annotated source
-
the goal is to identify the most likely protein in a mixture
-
a suitable reference database is available and correctly selected
-
a reference sequence already exists
-
the goal is confirmation, variant detection, or QC documentation
-
coverage against an expected sequence is the main deliverable
-
only N-terminal or C-terminal information is needed
-
the question is boundary confirmation rather than full-length assembly
Introduction
Protein sequence analysis projects rarely begin with a method already chosen. More often, a team starts with a sample and a decision to make: confirm a recombinant product, identify an unknown band, recover an antibody sequence, or document primary structure for publication or QC release. The same LC-MS/MS platform may support multiple workflows, but the best analytical route depends on whether a reliable reference sequence exists.
Researchers commonly compare de novo sequencing, database-assisted protein identification, peptide mapping, and terminal sequencing methods such as Edman degradation. Each approach can produce useful data. Each also has a distinct scope. Choosing the wrong one can waste time, increase cost, and still leave the project without the evidence needed for the next step.
The central decision is not which method is "better" in general. It is which method best matches the sample, the reference information available, and the level of evidence required. If your team is deciding between database-free sequence recovery and reference-based confirmation, MtoZ Biolabs can compare methods before samples are prepared or submitted.
Common Decision Scenarios
Method selection usually starts with one of four scenarios:
1. Unknown protein or missing reference.
A purified protein or gel band must be sequenced, but no trustworthy database entry exists.
2. Recombinant product confirmation.
A reference sequence is available, and the goal is to verify that the expressed product matches the intended design.
3. Terminal sequence question.
The project requires N-terminal or C-terminal confirmation rather than full-length sequence recovery.
4. Primary structure documentation for QC or publication.
The team needs traceable MS evidence, coverage maps, and report-ready deliverables.
These scenarios overlap, but they lead to different method priorities. Unknown proteins push teams toward database-free sequencing. Reference-backed QC projects often favor peptide mapping or database-assisted identification.
Related Services
| Customer Need | Recommended Service Direction |
|---|---|
| Want to confirm purified protein identity | |
| Want to confirm if the N-terminal or C-terminal is correct | / |
| Want to verify recombinant protein sequence coverage | |
| No reliable database sequence | |
| Want to analyze truncation, modification, or processing events |
Key Comparison Dimensions
A useful comparison should focus on decision-relevant factors rather than instrument brand or generic marketing claims. Four dimensions matter most:
The table below summarizes how de novo sequencing and database-assisted MS differ across these dimensions.
| Comparison Dimension | De Novo Sequencing | Database-Assisted MS |
|---|---|---|
| Reference sequence required | No | Yes |
| Best for unknown proteins | Strong fit | Limited fit |
| Best for sequence confirmation | Moderate fit | Strong fit |
| Expert interpretation need | High | Moderate |
| Typical turnaround | Often longer | Often faster |
| Ideal deliverable | Assembled sequence from spectra | Peptide coverage against reference |
De Novo Sequencing
The method interprets MS/MS fragment ions directly and assembles peptide sequences without relying on a database match. It is the preferred route when the correct reference is absent, proprietary, incomplete, or clearly inconsistent with the experimental data.
Strengths include database independence, direct protein-level evidence, and support for unknown protein analysis, antibody sequence recovery, and proprietary construct verification. The method is also valuable when database search returns low-confidence results despite good spectral quality.
Limitations include higher interpretation complexity, dependence on peptide coverage, and greater difficulty in repetitive or homologous regions. Database-free sequencing is usually not the fastest or lowest-cost option when a valid reference already exists.
For full-length unknown proteins, teams may review options. For shorter peptide targets, may be more appropriate.
Database-Assisted Protein Identification
Database-assisted MS matches experimental spectra to entries in UniProt, RefSeq, or custom sequence databases. It is efficient when the true sequence is already represented in the selected database and the sample quality supports confident peptide-spectrum matching.
This approach is widely used for protein identification from gel bands, pull-down samples, and complex mixtures when the goal is to name the most likely protein candidates. It works best when the biological context and database selection are correct.
The main limitation is dependence on reference completeness. If the sequence is missing or incorrect, the search engine may return weak matches, unrelated proteins, or misleading identifications. In those cases, repeating the same database workflow is rarely sufficient.
Peptide Mapping and Terminal Sequencing
Peptide mapping is often the best choice when a reference sequence already exists and the project goal is confirmation rather than discovery. LC-MS/MS peptide mapping can verify sequence coverage, detect expected peptides, identify variants, and support for biopharmaceutical or QC-focused projects.
Terminal sequencing methods answer a narrower question. and are useful when the project requires boundary confirmation, blocked termini checks, or limited terminal evidence rather than full-length recovery.
Edman degradation remains useful for specific N-terminal questions, but it is not typically a substitute for full de novo protein sequencing when complete primary structure is required.
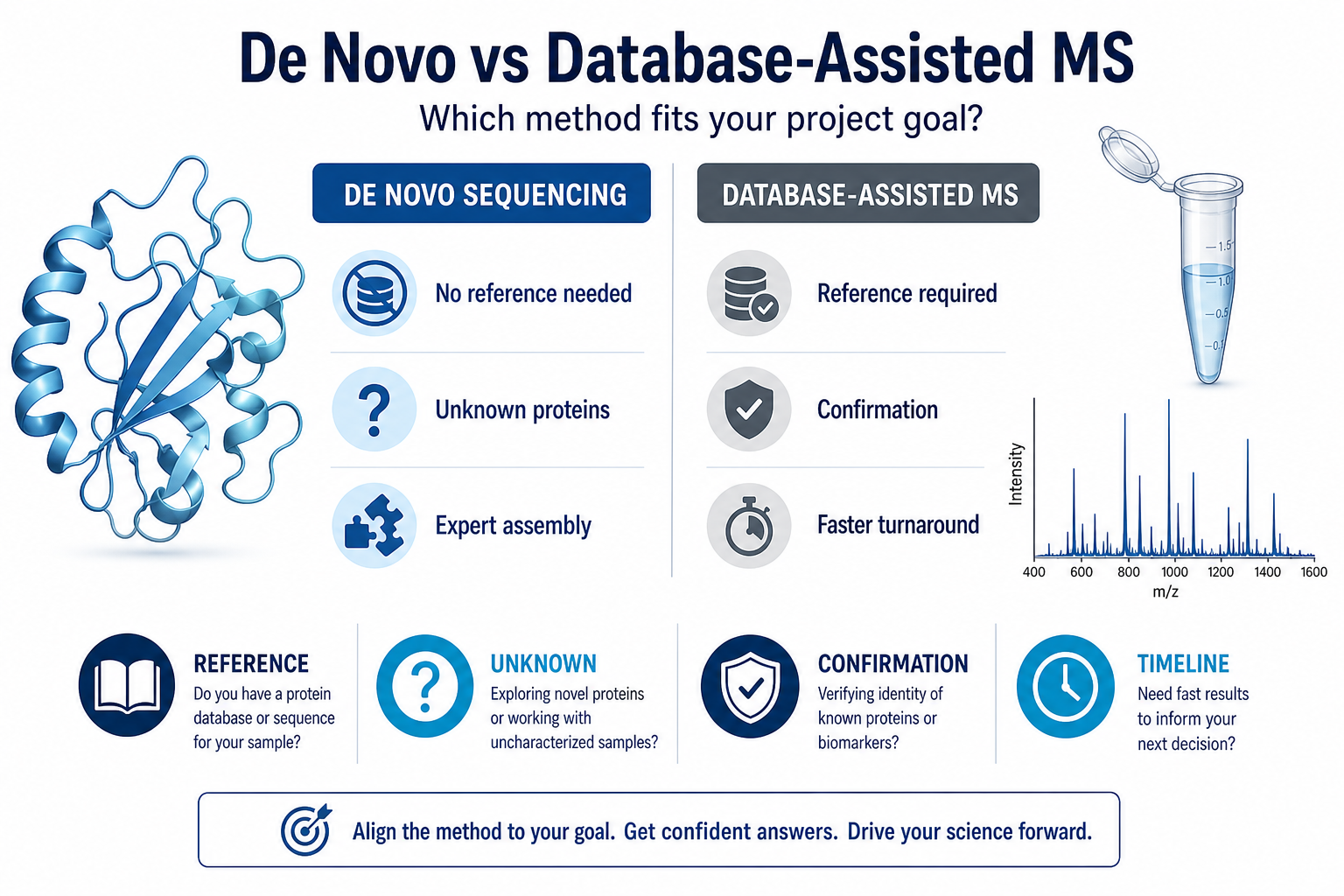
Figure 1. Method choice depends on reference availability and project goal.
Decision Recommendations by Project Goal
Use the following rules as a practical starting point:
Choose database-free sequencing when:
Choose database-assisted identification when:
Choose peptide mapping when:
Choose terminal sequencing when:
In-House vs Outsourced Sequence Analysis
Some organizations consider building internal LC-MS/MS sequence capability. This can make sense for large, recurring protein characterization programs with existing instrument infrastructure and dedicated interpretation staff. However, sequence analysis requires more than instrument access. It also requires digestion strategy, method development, QC review, reporting standards, and project management.
Outsourcing can reduce setup time and provide access to specialized de novo sequencing workflows for difficult or occasional projects. The tradeoff is vendor dependence, so teams should evaluate deliverables, communication, data ownership, and documentation quality before selecting a partner.
For many academic labs and biotech teams, outsourced support is most valuable when the project is urgent, sample-limited, documentation-sensitive, or outside routine internal capability.
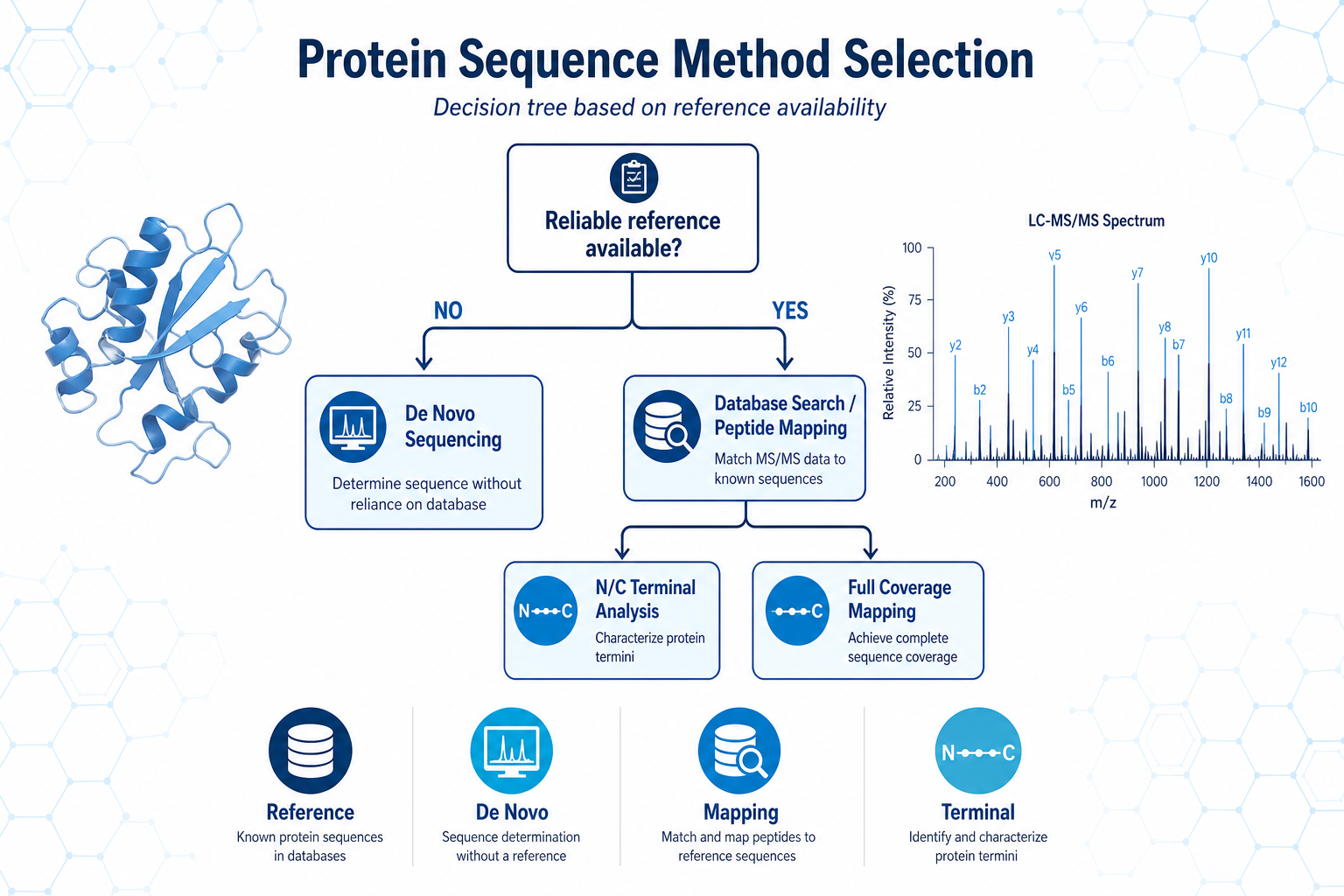
Figure 2. Reference availability is the first branch point in protein sequence method selection.
Frequently Asked Questions
1. Is de novo sequencing always better than database search?
No. Database-assisted MS is often faster and more efficient when a reliable reference exists. This approach is most valuable when the reference is missing or untrustworthy.
2. Can I use peptide mapping and de novo sequencing together?
Yes. Some projects begin with de novo recovery of unknown regions and later use peptide mapping for confirmation once a reference sequence is established.
3. When should I choose terminal sequencing instead of de novo sequencing?
Terminal sequencing fits projects that need N-terminal or C-terminal confirmation only. Full-length unknown sequence recovery usually requires LC-MS/MS-based de novo sequencing.
4. Does sample purity affect method choice?
Yes. Pure samples support stronger peptide coverage and clearer interpretation for both de novo sequencing and database-assisted workflows. Mixed samples may require additional purification before any sequence method is reliable.
5. Can database-assisted MS identify proprietary proteins?
Only if the proprietary sequence is included in the search database. Otherwise, database-assisted identification will fail or return incorrect matches.
Conclusion
Database-free sequencing and database-assisted MS answer different sequence questions. Database-assisted workflows are efficient for identification and confirmation when a valid reference exists. Database-free sequencing is the stronger choice for unknown proteins, failed database search recovery, and database-free primary structure determination. Peptide mapping and terminal sequencing remain valuable when the project scope is confirmation rather than discovery.
Method selection should begin with reference availability, sample quality, and the evidence standard required for the next decision. MtoZ Biolabs can match the workflow to sample type and project goal across , , and
How to order?

