





De Novo Sequencing: How LC-MS/MS Determines Protein Sequences Without a Database
-
determine the sequence of an unknown or novel protein
-
confirm a recombinant expression product
-
recover sequence information from purified antibody or peptide material
-
document primary structure for manuscripts or QC files
-
investigate truncation, processing, or sequence variants
-
sample feasibility review and purity assessment
-
enzymatic digestion, often with multiple proteases
-
LC separation and high-resolution MS/MS acquisition
-
de novo interpretation of peptide spectra
-
sequence assembly from overlapping peptides
-
expert review, coverage mapping, and report delivery
-
assembled amino acid sequence or peptide sequence calls
-
peptide coverage map across the protein
-
annotated MS/MS spectra supporting key sequence regions
-
notes on ambiguous residues, modifications, or low-confidence segments
-
QC summary and method description
-
raw spectral data when required for publication or audit review
Introduction
Protein sequence information is often required before a research team can move forward with expression, functional validation, publication, or quality documentation. In many projects, however, no reliable reference sequence exists. The protein may come from an unannotated organism, a proprietary expression system, a legacy purified sample, or a recombinant product that has not yet been fully verified.
Standard database-assisted mass spectrometry depends on matching experimental spectra to known sequences in UniProt or other repositories. When the correct sequence is missing, incomplete, or incorrect, database search alone cannot produce a trustworthy answer. Low-confidence peptide-spectrum matches, poor coverage, and ambiguous protein assignments are common warning signs.
De novo sequencing addresses this gap by interpreting tandem mass spectra directly from fragment ion patterns, without requiring a prior sequence match. The method is widely used for unknown proteins, antibody sequence recovery, peptide identification, and primary structure confirmation when database search is not sufficient. For teams evaluating whether a sample can support database-free sequence analysis, MtoZ Biolabs can review project fit before sample preparation begins.
What Question Does De Novo Sequencing Answer?
At its core, this method answers a practical question: what amino acid sequence best explains the observed peptide fragmentation data from this sample?
This differs from protein identification by database search. Database-assisted workflows ask whether an existing entry matches the data. This approach asks what sequence the data itself supports, even when no suitable reference is available.
The method is especially relevant when researchers need to:
Database-free sequencing does not replace every sequence workflow. When a reliable reference exists and the goal is confirmation, or database-assisted may be more efficient. Database-free analysis becomes essential when the reference itself is the unknown.
Related Services
| Customer Need | Recommended Service Direction |
|---|---|
| Want to confirm purified protein identity | |
| Want to confirm if the N-terminal or C-terminal is correct | / |
| Want to verify recombinant protein sequence coverage | |
| No reliable database sequence | |
| Want to analyze truncation, modification, or processing events |
How De Novo Sequencing Works
The workflow is built on LC-MS/MS analysis of enzymatically digested peptides. A purified protein or peptide sample is digested with one or more proteases to generate overlapping peptide fragments. Peptides are separated by liquid chromatography and analyzed by high-resolution tandem mass spectrometry.
During MS/MS, peptide ions are fragmented in the mass spectrometer. The resulting product ions, commonly b-ions and y-ions, reflect cleavage between amino acid residues. Software and expert analysts use these fragment patterns to infer short sequence tags and then assemble longer sequences through overlapping peptide evidence.
A typical LC-MS/MS sequencing workflow includes:
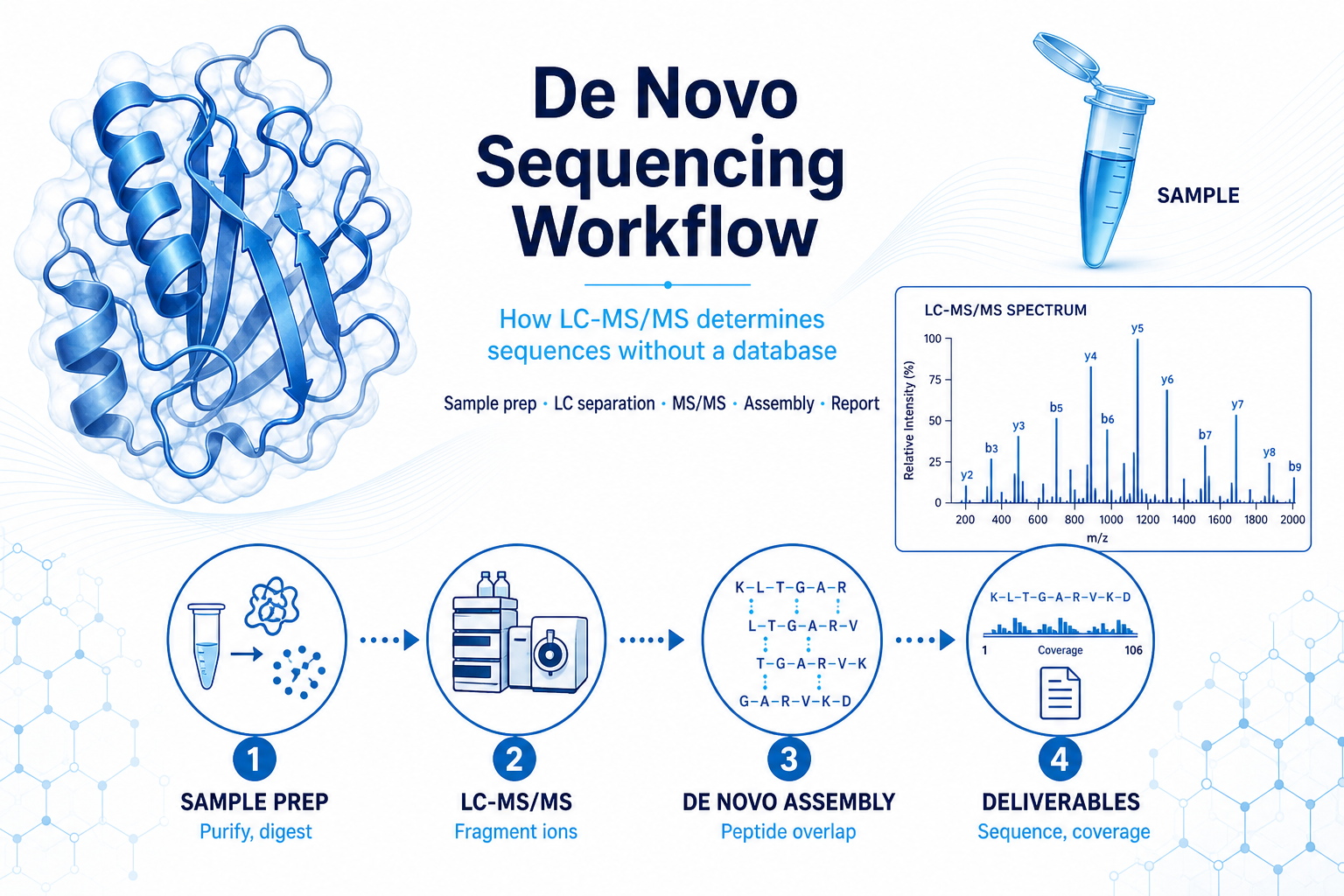
Figure 1. De novo sequencing converts purified protein into sequence evidence through digestion, LC-MS/MS, and expert assembly.
The quality of the final sequence depends on peptide coverage, spectral quality, digestion strategy, and the complexity of the sample. For this reason, de novo protein sequencing is not simply an automated search task. Expert interpretation remains important, especially for long proteins, modified peptides, or samples with homologous regions.
Core Advantages and Current Limitations
Core Advantages
Database independence. This method can proceed when no suitable reference sequence exists in public or internal databases.
Direct protein-level evidence. The method works from purified protein or peptide material, which is valuable for legacy samples, commercial proteins, and antibody products without recoverable genetic material.
Support for discovery and confirmation. The approach can identify unknown proteins, verify recombinant products, and provide primary structure evidence for downstream expression or regulatory use.
Compatibility with orthogonal follow-up. Sequence results can be supported by , , or when additional confirmation is needed.
Current Limitations
Coverage dependent. Incomplete digestion, low sample amount, or poor spectral quality can leave gaps in the assembled sequence.
Ambiguity in repetitive or homologous regions. Similar amino acid motifs, isobaric residues, and highly homologous sequences can complicate assembly.
Modified peptides require careful interpretation. Post-translational modifications, chemical adducts, and missed cleavages can affect fragment interpretation.
Not every project needs full de novo sequencing. If a reliable reference exists, database-assisted confirmation is often faster and more cost-effective.
Typical Application Scenarios
De novo sequencing is most useful when sequence identity is biologically or operationally critical, but not available from existing records.
Researchers may consider database-free sequencing in the following situations:
1. Unknown or novel proteins. A band on SDS-PAGE or a purified fraction may require sequence determination before cloning, expression, or functional study.
2. Recombinant protein verification. A production batch may need protein-level confirmation that the expressed product matches the intended design.
3. Antibody or peptide samples without genetic source. Legacy antibodies, commercial reagents, or purified peptide material may lack accessible DNA or RNA.
4. Truncation or processing analysis. Signal peptide removal, C-terminal processing, or unexpected proteolysis may require direct protein sequence evidence.
5. Publication or QC documentation. Manuscripts, tech transfer files, and internal release testing may require primary structure data with traceable MS evidence.
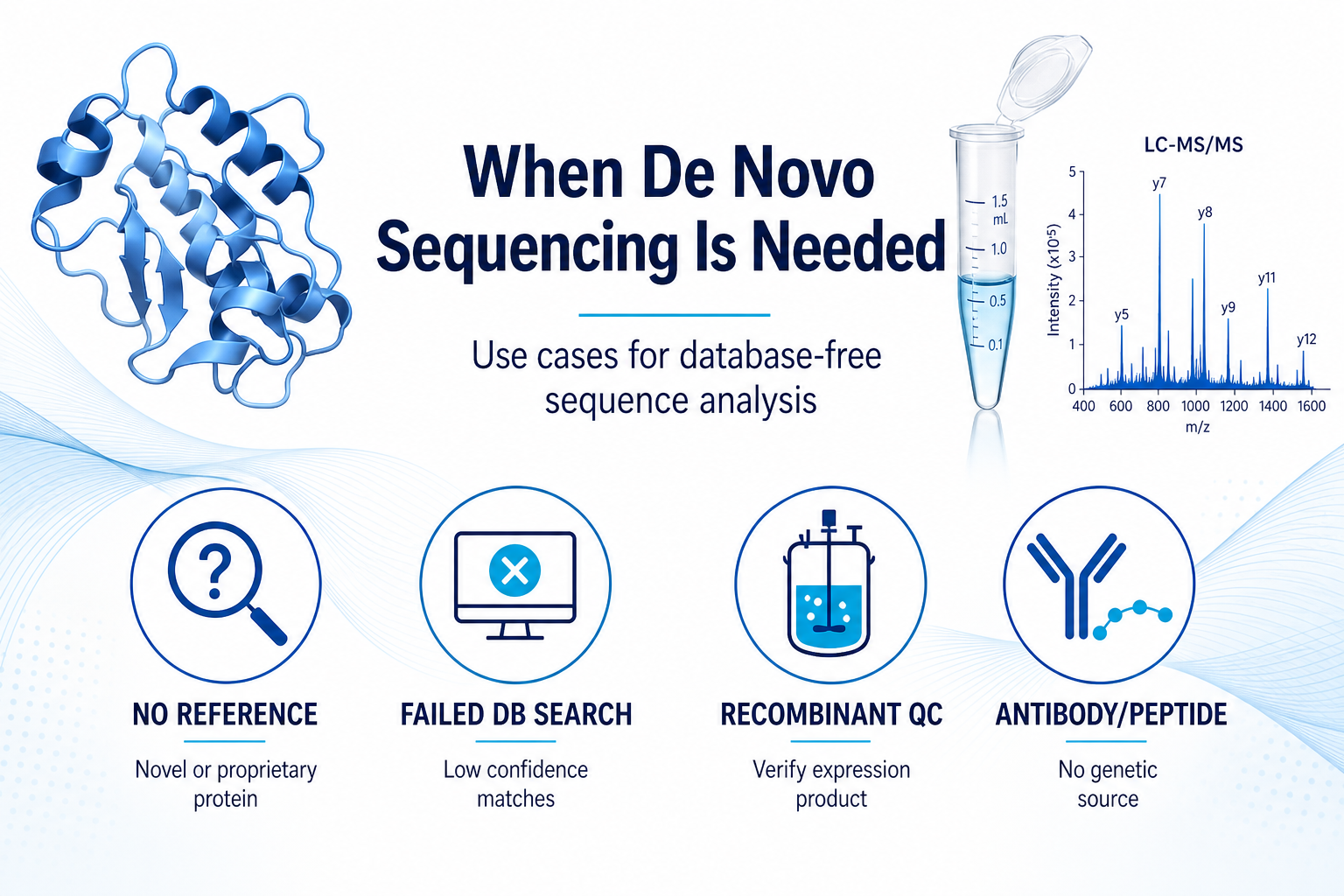
Figure 2. De novo sequencing is most valuable when reference sequence information is missing or unreliable.
For antibody-focused projects, teams may also review options when the sample is immunoglobulin rather than a general protein.
What Results Should Researchers Expect?
A strong sequence analysis report should connect the reported sequence to supporting evidence. Useful deliverables often include:
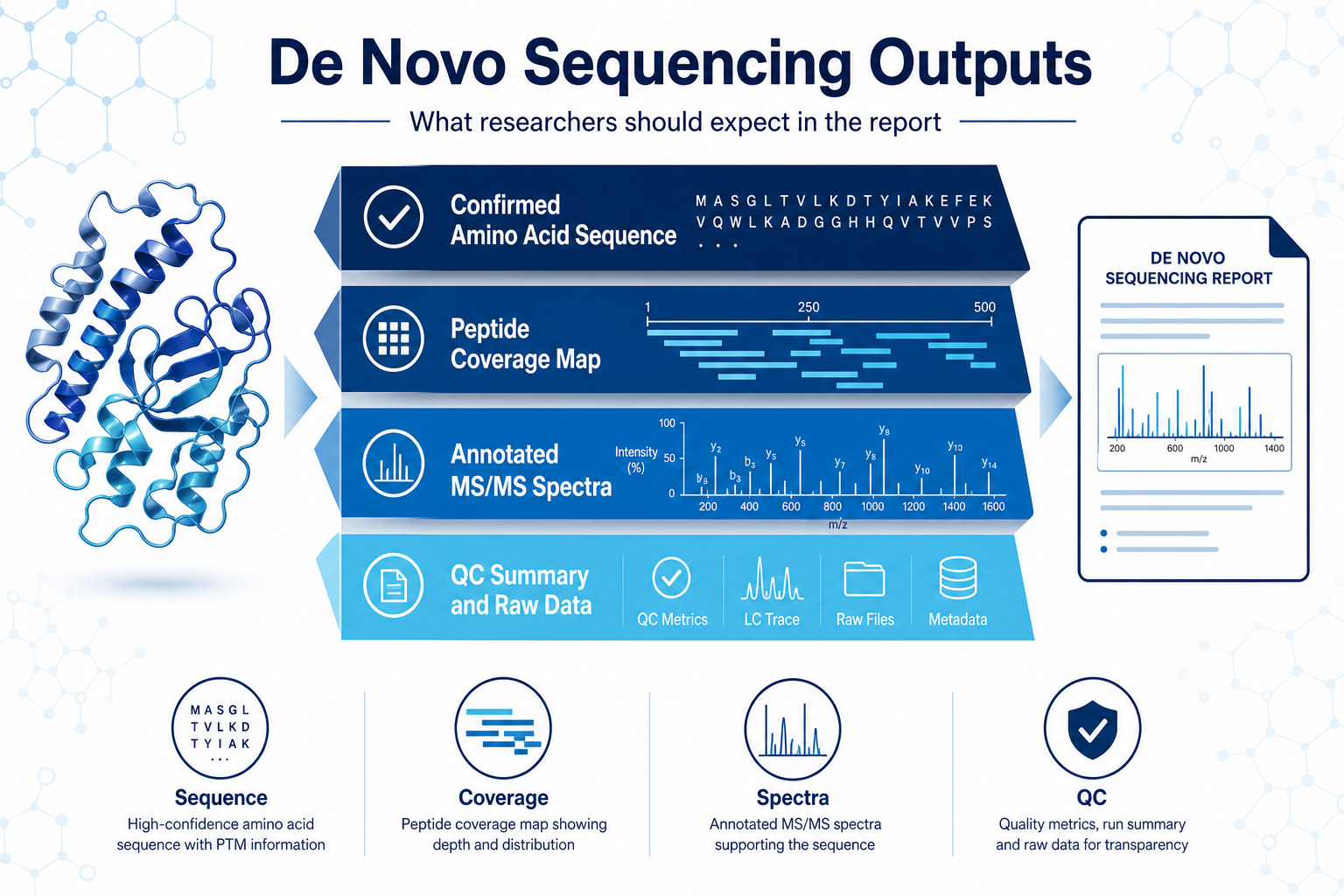
Figure 3. Publication-ready de novo sequencing deliverables should link sequence calls to supporting MS evidence.
Researchers should treat low-coverage regions cautiously. A partial sequence may still be valuable for cloning design or follow-up validation, but it should not be over-interpreted as complete primary structure evidence without additional coverage.
Frequently Asked Questions
1. Can de novo sequencing work without any reference sequence?
Yes. That is the main purpose of the method. The workflow interprets MS/MS spectra directly and assembles sequence information from peptide evidence, even when no database match is available.
2. Is de novo sequencing the same as protein identification?
No. Protein identification usually depends on matching spectra to known entries in a database. Database-free analysis builds the sequence from the spectra themselves.
3. Can de novo sequencing confirm a recombinant protein?
Yes. Many teams use database-free sequencing or to verify that an expressed product matches the intended design, especially when expression anomalies or processing events are suspected.
4. What sample types are commonly used?
Purified proteins, gel bands, recombinant products, peptides, and some antibody preparations are common starting materials. Sample purity, amount, and buffer composition strongly affect success.
5. Does de novo sequencing identify post-translational modifications?
PTMs can be detected in some cases, but PTM-focused projects may require dedicated or peptide mapping workflows with explicit modification search parameters.
Conclusion
This method provides a database-free route to protein and peptide sequence determination when reference information is missing, unreliable, or insufficient. By combining LC-MS/MS, overlapping peptide evidence, and expert interpretation, the method supports unknown protein analysis, recombinant verification, antibody sequence recovery, and primary structure documentation. It is not the best choice for every project, but it is often the most direct option when database search cannot answer the biological question.
For unknown proteins, low-confidence database results, or QC-driven sequence confirmation, MtoZ Biolabs provides support covering feasibility review, LC-MS/MS analysis, sequence assembly, and publication-friendly reporting. Researchers can contact the technical team to evaluate sample type, coverage expectations, and the best validation path before samples are submitted.
How to order?

