





De Novo Protein Sequencing for Unknown Proteins: Sample Prep and Coverage Optimization
-
only a small fraction of the protein sequence is supported by peptides
-
database search returns weak matches despite a clean-looking band
-
multiple protease digests still fail to cover difficult regions
-
the sample appears pure by SDS-PAGE but produces mixed peptide evidence
-
the protein is large, modified, or repetitive, making assembly ambiguous
-
the project deadline requires full-length sequence evidence, not partial coverage
-
assembled protein sequence regions with confidence annotations
-
peptide coverage map showing supported and unsupported segments
-
annotated MS/MS spectra for major sequence calls
-
notes on ambiguous residues, modifications, or low-confidence areas
-
recommendations for follow-up validation if full coverage was not achieved
-
Intact mass measurement to confirm overall molecular weight consistency
-
N-terminal or C-terminal sequencing for boundary confirmation
-
Peptide mapping when a reference becomes available after initial protein-level recovery
-
Recombinant expression and functional testing when the sequence will be used for production
Introduction
A purified protein sample does not always translate into a complete sequence report. Researchers may submit a visible gel band or a chromatography fraction with confidence, yet receive only partial peptide coverage, ambiguous assembly, or a report that stops short of the full primary structure. For teams working on unknown proteins, recombinant QC, or legacy purified material, incomplete coverage is one of the most common reasons a protein sequencing project stalls.
Low coverage is rarely caused by a single factor. Sample purity, protein amount, buffer composition, digestion strategy, LC-MS/MS depth, and protein complexity all influence whether overlapping peptide evidence is strong enough for reliable assembly. Repeating the same submission without adjusting preparation or method design often produces the same incomplete result.
De novo protein sequencing can recover strong primary structure evidence when the workflow is matched to the sample and coverage goal. The key is to identify why coverage is weak before resubmitting material or expanding the analytical plan. If your team is troubleshooting low peptide coverage or preparing an unknown protein sample for the first time, MtoZ Biolabs can Assess sample readiness and recommend digestion, LC-MS/MS, and validation steps before sequencing begins.
Common Pain Points in Protein Sequencing Projects
Researchers often seek help after encountering one or more of the following issues:
These problems are common in unknown protein discovery, recombinant expression QC, enriched protein fractions, and legacy purified samples with incomplete metadata. In many cases, the issue is not whether protein sequencing is possible. It is whether the sample and method design can generate enough overlapping evidence for the required decision.
Why Protein Sequence Coverage Falls Short
Before changing methods, it helps to understand why peptide coverage may remain incomplete.
1. Sample Quality Issues
Mixed proteins, degradation, low amount, or incompatible buffer components can reduce the number of usable peptides before LC-MS/MS begins.
2. Suboptimal Digestion Strategy
A single protease may not cleave evenly across the protein, leaving long regions without useful peptides. Missed cleavages and resistant domains can also limit coverage.
3. Insufficient LC-MS/MS Depth
Weak fragmentation, low signal-to-noise ratios, or limited instrument time can reduce the number of high-quality spectra available for assembly.
4. Protein Complexity
Large size, repetitive motifs, homologous regions, and post-translational modifications can all make sequence assembly more difficult even when data are obtained.
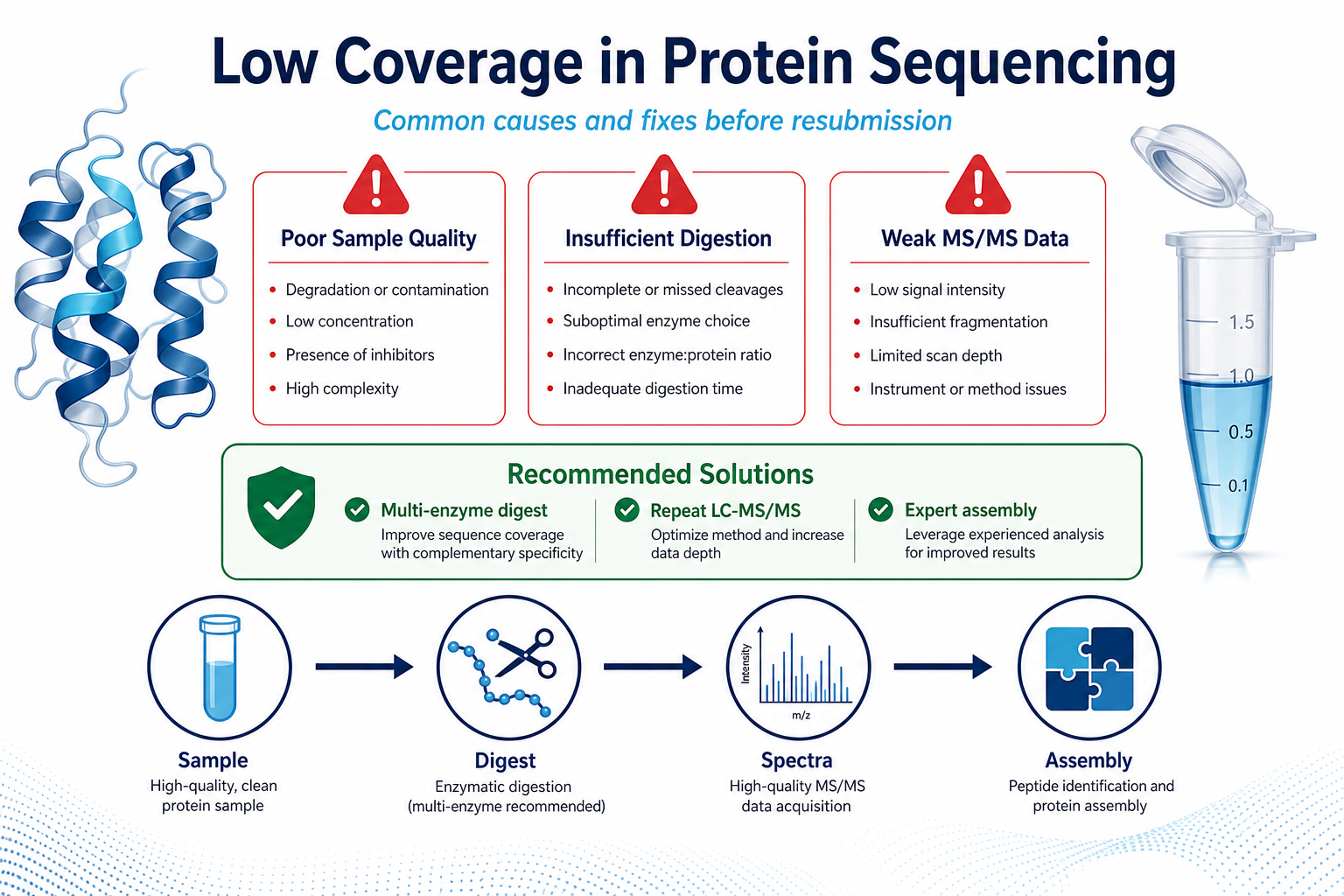
Figure 1. Low coverage often reflects sample quality, digestion design, or MS/MS depth rather than method failure alone.
Related Services
| Customer Need | Recommended Service Direction |
| Want to confirm purified protein identity | Protein Identification Service |
| Want to confirm if the N-terminal or C- terminal is correct | N-Terminal Sequencing Service / C-Terminal Sequencing Service |
| Want to verify recombinant protein sequence coverage | Peptide Mapping Service |
| No reliable database sequence | De Novo Protein Sequencing Service |
| Want to analyze truncation, modification, or processing events | Primary Structure Analysis Service |
Step-by-Step Coverage Optimization Guide
When peptide coverage is insufficient, use a structured review rather than repeating the same digestion and LC-MS/MS plan.
Step 1: Confirm Protein Purity and Integrity
Review SDS-PAGE, staining intensity, and sample handling history. A dominant band does not always mean a single protein. Degraded or partially proteolysed material can reduce usable peptide evidence.
Step 2: Evaluate Sample Amount and Buffer Compatibility
Confirm that enough material is available for multi-enzyme digestion and repeat LC-MS/MS if needed. High salt, strong detergents, and interfering additives may require cleanup before digestion.
Step 3: Redesign the Digestion Strategy
Use complementary proteases to increase overlapping peptide coverage. Trypsin, chymotrypsin, Glu-C, and other enzymes can expose different regions of the protein and improve assembly potential.
Step 4: Increase LC-MS/MS Depth
If initial spectra are weak or sparse, additional instrument time, fractionation, or repeat analysis may be required. Coverage optimization often depends on obtaining more high-quality MS/MS spectra, not simply rerunning the same method.
Step 5: Plan Validation for Unsupported Regions
If full-length coverage is not achieved, define which regions are supported and whether terminal sequencing, intact mass measurement, or targeted follow-up is needed for the next project decision.
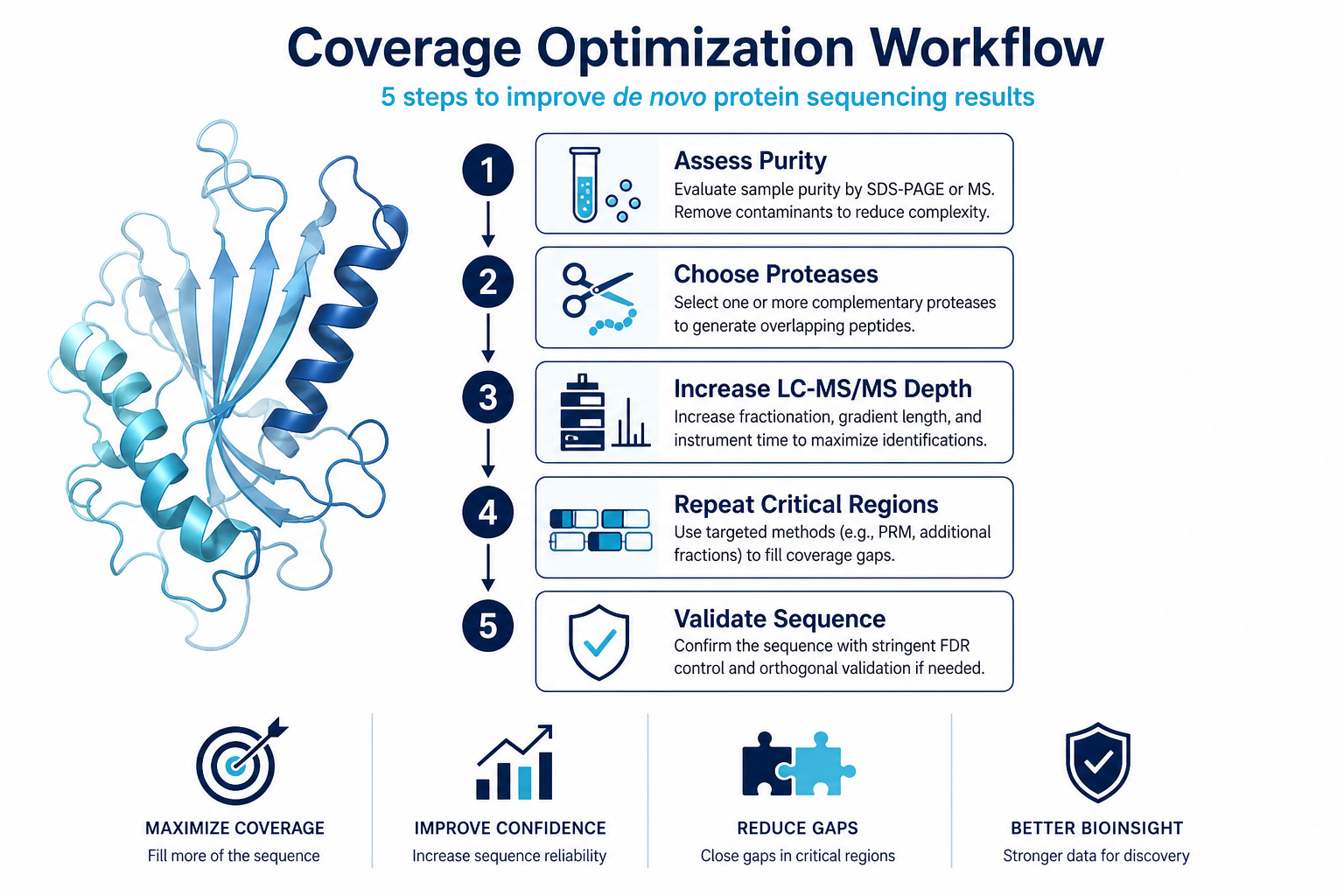
Figure 2. A structured optimization path reduces repeat submissions and improves sequence confidence.
Sample Preparation Best Practices
Sample preparation is often the highest-leverage step in a protein sequencing project. The figure below summarizes recommended practices and common risk factors.

Figure 3. Feasibility review before sample submission reduces rework and improves coverage outcomes.
For gel-based samples, excise the target band as precisely as possible and minimize contamination from neighboring bands. For recombinant proteins, document expression system, expected size, and purification method. For legacy samples, provide any historical information that may explain unusual processing or truncation.
Teams working with difficult unknown proteins may also consider Unknown Proteins Sequencing Service when the sample history is incomplete but the protein fraction is the only available material.
Expected Results and Validation Methods
A successful protein sequencing project should deliver more than a partial sequence string. Expected outputs may include:
Validation options depend on project goal:
Protein-level sequence recovery can provide strong primary structure evidence, but validation should match the decision the data must support.
Key Cautions
Do not assume that low coverage means the wrong method was chosen. Coverage problems often begin with sample preparation or digestion design.
Do not treat partial coverage as full sequence confirmation. Unsupported regions should be reported with appropriate caution.
Do not skip metadata. Accurate sample history helps the sequencing team choose proteases, interpret homologous regions, and avoid unnecessary repeat analysis.
For difficult proteins, a combined strategy may be best. De novo protein sequencing can recover unknown regions, while protein identification or peptide mapping can confirm regions once a reference is established.
Frequently Asked Questions
1. How much protein is needed for de novo protein sequencing?
Requirements depend on protein size, purity, and complexity. A feasibility review is recommended before submission, especially for low-abundance or gel-band samples.
2. Can a single protease digest provide full protein coverage?
Sometimes, but many projects benefit from multiple proteases to increase overlapping peptide evidence across the protein sequence.
3. Why does a clean gel band still produce incomplete coverage?
The band may contain multiple proteins, degraded material, or a protein with regions that are difficult to digest or fragment efficiently.
4. Can low coverage still support a project decision?
Yes, if the supported regions are clearly documented and sufficient for the next step, such as cloning design or targeted validation. Full-length claims require adequate coverage.
5. Should I purify the protein further before resubmitting?
Often yes. Additional purification can reduce mixed peptide evidence and improve assembly confidence.
Conclusion
Incomplete peptide coverage is one of the most common barriers in unknown protein sequencing projects, but it is often addressable through better sample preparation, digestion design, and LC- MS/MS depth planning. De novo protein sequencing can deliver strong primary structure evidence when the workflow is matched to the sample and the required level of coverage is defined early.
When low coverage or sample complexity is blocking progress, MtoZ Biolabs can Plan a coverage strategy using De Novo Protein Sequencing Service, terminal analysis, or peptide mapping based on sample type and project goal. Contact the technical team to review sample status, digestion options, and the fastest path to usable protein sequence evidence.
How to order?

