





Can Full-Length Protein Sequencing Detect Post-Translational Modifications
-
Mass spectrometry can infer the presence of modification moieties by detecting subtle mass differences among peptide fragments;
-
Certain PTMs generate characteristic fragmentation patterns (e.g., neutral loss for phosphorylation, dehydration peaks for glycosylation), which can assist in the identification of modification types;
-
However, PTMs that are low in abundance, structurally atypical, or lacking signature fragment ions remain difficult to detect directly.
-
Antibody Drug Development: For example, N-terminal acetylation of the antibody light chain may influence its structural stability and immunogenicity.
-
Validation of Recombinant Proteins: Determining whether aberrant modifications, such as abnormal glycosylation or oxidative damage, occur during expression is crucial for quality control.
-
Signaling Pathway Analysis: Mapping phosphorylation or ubiquitination sites on functional proteins aids in elucidating their roles in cellular regulatory mechanisms.
-
Clinical Biomarker Discovery: PTM sites associated with disease states can serve as valuable diagnostic and prognostic indicators.
Post-translational modifications (PTMs) of proteins represent one of the central mechanisms underlying protein diversity and functional regulation. Modifications such as phosphorylation, acetylation, glycosylation, hydroxylation, and ubiquitination play essential roles in signal transduction, immune modulation, and metabolic control. With the rapid advancement of full-length protein sequencing technologies and associated de novo interpretation algorithms, increasing attention has been directed toward a key question: is it possible to identify post-translational modifications directly from spectral data, without relying on database matching? The answer is: some PTMs can indeed be detected; however, technical dependencies and limitations in modification recognition still exist. This article will explore the issue from four perspectives: technical principles, identifiable PTM types, methodological integration, and future application prospects.
Principle of Full-Length Protein Sequencing: Can Modification Information Be "Observed"?
Full-length protein sequencing typically relies on high-resolution mass spectrometry platforms (e.g., Orbitrap, TOF, FT-ICR), and employs multiple enzymatic digestion strategies, spectral reconstruction, and de novo sequence inference to determine the complete amino acid sequence from the N-terminus to the C-terminus. In this process:
Therefore, while protein full-length sequencing is theoretically capable of identifying PTMs, its accuracy is contingent upon the performance of the mass spectrometry instrumentation, the chosen fragmentation strategy, and the data analysis algorithms employed.
Common Identifiable Types of Post-Translational Modifications
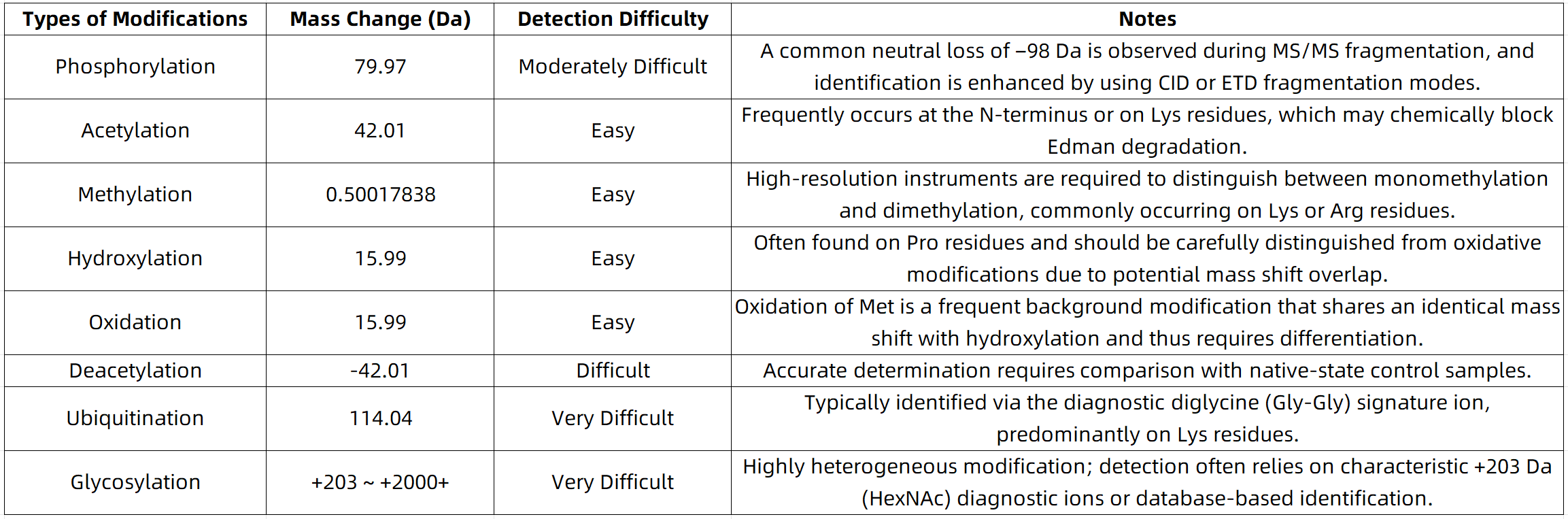
In practical sequencing services, common modifications such as phosphorylation, acetylation, and oxidation are often reliably identified. However, more complex modifications, including glycosylation and ubiquitination, typically require dedicated enrichment strategies or auxiliary analysis with specialized databases to achieve accurate detection.
Strategies to Enhance PTM Detection Accuracy
1. Employing High-Resolution, High-Sensitivity Mass Spectrometry Platforms
Advanced instruments such as Orbitrap Fusion Lumos, timsTOF Pro, and Q Exactive HF-X provide high mass accuracy at the parts-per-million (ppm) level and rapid scan rates, enabling the differentiation of subtle mass shifts induced by post-translational modifications.
2. Multi-Enzyme Digestion Approaches
Single-protease digestion strategies may fail to reveal certain modification sites. By employing a combination of enzymes such as Trypsin, Chymotrypsin, Glu-C, and Lys-C, the overall sequence coverage can be significantly increased, thereby uncovering a broader range of potential PTM sites.
3. Application of Gentle Fragmentation Techniques, such as ETD/EThcD
In contrast to collision-induced dissociation (CID), techniques like electron-transfer dissociation (ETD) and electron-transfer/higher-energy collision dissociation (EThcD) preserve labile modifications. These approaches are particularly suitable for identifying unstable PTMs such as phosphorylation and glycosylation.
4. Optimization of Data Analysis Algorithms
The use of advanced software platforms including Byonic, PEAKS Studio, pNovo, and MetaMorpheus, which support PTM discovery, allows for the identification of modification types and localization even in the absence of reference protein sequences.
Applications: Why Is PTM Detection So Critical?
Accurate identification of PTMs plays a pivotal role in both scientific research and biopharmaceutical development:
MtoZ Biolabs’ protein full-length sequencing service integrates de novo peptide sequencing with robust PTM identification capabilities. This is supported by multi-enzyme digestion protocols and high-resolution mass spectrometry platforms, and all results undergo manual validation by experienced specialists to ensure both accuracy and reliability in PTM detection.
Summary: Full-Length Protein Sequencing Enables PTM Detection with the Aid of Complementary Strategies
Full-length protein sequencing is capable of detecting a wide range of post-translational modifications, particularly those characterized by distinct mass shifts and identifiable fragmentation patterns. However, the reliable detection of low-abundance, structurally complex, or spectrally ambiguous PTMs remains challenging. Improvements in sample preparation, high-resolution instrumentation, tailored fragmentation methods, and optimized computational algorithms are essential for enhancing detection sensitivity and specificity. As protein modification analysis transitions from database-dependent approaches to direct interpretation of raw spectral data, MtoZ Biolabs stands at the forefront as a leading enabler of this technological evolution.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

