





Subcellular Proteomics Workflow (Including Mass Spectrometry and SILAC Approaches)
-
Reveal dynamic trafficking of proteins among distinct organelles.
-
Identify disease-associated, compartment-specific abnormalities in protein expression.
-
Reconstruct protein network organization across spatiotemporal contexts.
-
Differential centrifugation: applicable to coarse enrichment of many organelles (e.g., mitochondria, nuclei, vesicles).
-
Density gradient centrifugation (e.g., sucrose or Percoll gradients): used for further purification of the target organelle.
-
Immunoaffinity isolation: antibody-based enrichment of specific organelles, suitable for cellular structures that are difficult to separate by centrifugation.
-
Subcellular Localization Mapping: combined with technologies such as SILAC to enhance spatial resolving power.
-
For membrane-protein-enriched samples, a high-salt buffer combined with non-ionic detergents is recommended.
-
For nuclear fractions containing nucleic acids, nucleases should be included to reduce sample viscosity.
-
No chemical derivatization is required, minimizing the risk of introducing non-physiological alterations.
-
Well suited for studies of dynamic trafficking.
-
Intrinsically compatible with mass spectrometry, offering high quantification accuracy.
-
Protein localization map construction: different SILAC-labeled conditions can be used to support localization inference based on label intensity and distribution patterns across subcellular fractions.
-
Dynamic trafficking studies: SILAC ratio changes before and after perturbation can reveal trends in protein redistribution.
-
Omics-scale comparisons under high-throughput screening settings.
-
Raw data preprocessing (Raw to mzML).
-
Isotope label identification and quantification (e.g., MaxQuant, Proteome Discoverer).
-
Support from subcellular localization inference tools (e.g., MetaMass, SubCons).
-
Enrichment analysis and functional annotation (GO, KEGG, Reactome).
-
Whether protein coverage is balanced across fractions.
-
Whether isotopic peak resolution is sufficient.
-
Whether batch effects are present and confound interpretation.
In the post-genomic era, subcellular proteomics has increasingly emerged as an important research direction for elucidating cellular function, signaling pathways, and disease mechanisms. In contrast to whole-cell proteomics, subcellular investigations focus on protein expression, localization, and dynamic changes within specific organelles (e.g., mitochondria, the endoplasmic reticulum, and the nucleus), thereby enabling higher spatial resolution proteome profiling.
Why Choose Subcellular Proteomics?
Although conventional proteomics has enabled substantial progress in disease biomarker discovery and signaling pathway studies, its spatial resolution is often limited. For instance, in tumor cells, whole-proteome expression changes do not necessarily capture organelle-level protein relocalization (protein translocation).
By enabling precise organelle isolation and quantitative analysis, subcellular proteomics can:
Within this research area, the integrated use of high-resolution mass spectrometry platforms and quantitative labeling strategies is particularly important.
Core Workflow of Subcellular Proteomics
1. Sample Preparation and Subcellular Fractionation
High-quality subcellular fractionation is the foundation of the entire workflow. Commonly used approaches include:
2. Protein Extraction and Enzymatic Digestion
Because protein composition and abundance vary substantially across organelles, extraction conditions require buffer optimization, for example:
Proteolytic digestion is typically performed using trypsin; when needed, auxiliary proteases such as Lys-C can be combined to improve cleavage efficiency for complex protein mixtures.
Detailed Quantification Strategies: Advantages of SILAC in Subcellular Proteomics
1. What Is SILAC?
SILAC (Stable Isotope Labeling by Amino acids in Cell culture) is a metabolic labeling strategy that introduces amino acids containing heavy isotopes during cell culture to achieve relative protein-level quantification. Key advantages include:
In subcellular proteomics, SILAC is particularly suitable for:
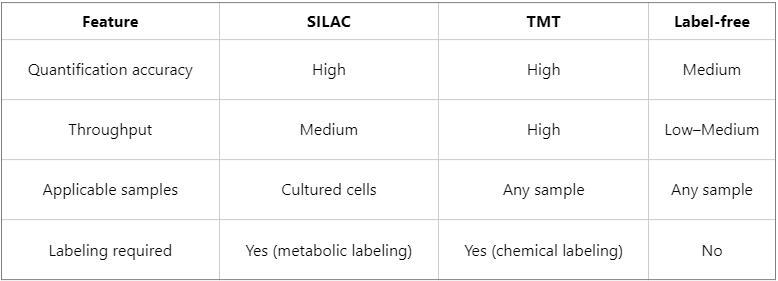
2. SILAC vs Other Quantification Technologies (TMT, Label-Free)
Mass Spectrometry Analysis and Data Interpretation
1. Instrument Selection: High Resolution vs High Throughput
For subcellular proteomics, high-resolution mass spectrometers (e.g., Orbitrap Exploris instruments and timsTOF-series platforms) are commonly recommended to improve detection of low-abundance proteins and isotopic labels.
2. Data Processing Workflow
A typical analytical workflow includes:
In practical projects, rigorous data quality control is particularly critical. For example:
Subcellular proteomics represents an important bridge between protein expression and cellular function. With continued improvements in mass spectrometry sensitivity and the expanding repertoire of quantitative strategies, high-precision subcellular omics has become indispensable for understanding complex biological phenomena. Looking ahead, emerging directions, including spatial multi-omics, AI-assisted localization prediction, and single-cell subcellular omics, are expected to further extend the boundaries of research. MtoZ Biolabs will continue to optimize an integrated workflow spanning sample processing, mass spectrometry analysis, and bioinformatics interpretation, thereby supporting researchers in exploring biological systems at higher resolution.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

