





Small Molecule Drug Screening Service-Receptor Categories
Receptors are biomolecules that can bind with hormones, neurotransmitters, drugs, or intracellular signaling molecules and are capable of inducing changes in cellular function. Receptors are categorized based on their cellular location into cell surface receptors and intracellular receptors. Each receptor typically has two active sites: one that recognizes and binds to the ligand and another functional active site that triggers a response. This response only occurs after the ligand binds to form a binary complex, causing a conformational change, initiating a series of biochemical reactions, and ultimately leading to a biological effect in the target cell.
Targeting receptor proteins, such as ligand-gated ion channels (LGICs) and G protein-coupled receptors (GPCRs), are crucial for the discovery of most drugs regulating receptor signaling pathways. GPCRs and LGICs are among the most significant targets for drugs, serving as the sites of action for nearly 40% of all approved drugs.
1. GPCRs
GPCRs constitute the largest protein family encoded by the human genome. They reside on cell membranes, translating extracellular signals into critical physiological responses. Their endogenous ligands span a diverse range, including odors, hormones, neurotransmitters, and chemotactic factors, etc., extending from photons, amines, carbohydrates, lipids, peptides to proteins. GPCRs are implicated in numerous diseases, such as type 2 diabetes mellitus (T2DM), obesity, depression, cancer, Alzheimer's disease (AD), and many others. Traditionally, the extracellular structures of GPCRs have been recognized as sites for ligand initiating GPCR-mediated intracellular signal transduction. This process involves the conformational change and subsequent activation of heterotrimeric G proteins, leading to the GDP-to-GTP exchange on the Gα subunit and its dissociation from the Gβγ subunits. The Gα subunit's subsequent hydrolysis of GTP to GDP, followed by reassociation with Gβγ, terminates G protein signaling, a process facilitated by RGS proteins. However, emerging research indicates that GPCRs also engage in various extracellular interactions. These complexes are highly diverse, potentially forming connections or synapse-specific environments that endow GPCRs with distinct functional characteristics.
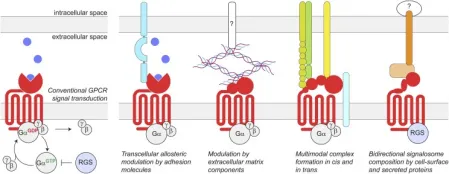
Figure 1. Schematic of Conventional GPCR Signal Transduction and Transcellular GPCR Complexes [6]
Approximately 350 of the 826 human GPCRs are considered druggable, with 165 being validated drug targets. Ongoing research into GPCR-targeting drugs predominantly involves exogenous small molecules, such as synthetic organics, natural products, and inorganics, which collectively represent about 64% of all researches.
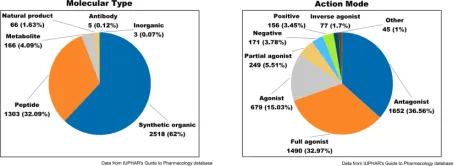
Figure 2. Drug Molecules Targeting GPCRs: Analysis of Molecular Types (Left) and Modes of Action (Right) [7]
Based on amino acid sequences, the human GPCR family is categorized into Class A (rhodopsin), Class B (secretin and adhesion), Class C (glutamate), and Class F (Frizzled) subfamilies. Class A GPCRs comprise a rhodopsin-like receptor family of 719 members, subdivided into aminegic, peptide, protein, lipid, melatonin, nucleotide, steroid, alicarboxylic acid, sensory, and orphan subclasses. These receptors feature a conventional transmembrane domain (TMD) that forms a ligand-binding pocket and includes 8 additional helices and palmitoylated cysteines at the C-terminal. Given their extensive physiological roles, this class represents the most targeted receptor type for therapeutic interventions. Class B GPCRs are bifurcated into two subfamilies: secretin (B1) and adhesion (B2), comprising 15 and 33 members, respectively. Secretin subfamily members possess an expanded extracellular domain (ECD), interacting with vasoactive intestinal peptide (VIP), pituitary adenylate cyclase-activating peptide (PACAP), corticotropin-releasing factor (CRF), parathyroid peptide hormone (PTH), growth hormone-releasing hormone (GHRH), calcitonin gene-related peptide (CGRP), glucagon, and glucagon-like peptides (GLPs). The adhesion receptor subfamily, comprising 9 subclasses, is characterized by distinct N-terminal motifs such as epidermal growth factor, cadherin, and immunoglobulin domains, setting them apart in their involvement in cell adhesion and migration. The C class glutamate receptors consist of 22 receptors, which are divided into 5 subfamilies, including 1 calcium-sensing receptor (CaSR), 2 γ-aminobutyric acid (GABA) B type receptors (GABAB1 and GABAB2), 3 taste 1 receptors (TS1R1-3), 8 metabotropic glutamate receptors (mGluR1-8), and 8 orphan GPCRs. A salient feature of the glutamate receptor family is its ECD and the obligated constitutive dimer for receptor activation. The F class GPCRs, identified as drug targets, include smooth receptors SMO with approved small molecule antagonists for cancer treatment. The other ten members, Frizzled receptors (FZD1-10), play a pivotal role in mediating Wnt signaling, indispensable for the development of embryo and adult.
The identification of compounds targeting receptor is contingent upon the target site, specifically whether it is extracellular, intramembrane, or intracellular. Antibodies and related biologics are particularly suited for binding to extensive surfaces, exhibiting high potency and specificity; hence, they are utilized for targeting subsets of receptor. However, their inability to readily traverse cell membranes or the blood-brain barrier significantly restricts their use. In contrast, small molecules are capable of cellular penetration and can target receptor complexes across all three categories. Nonetheless, small molecules are limited to covering modest surface areas, rendering them most effective in confined binding sites. Additionally, peptides and peptide derivatives present compelling alternatives for targeting both extracellular and intracellular receptors, despite their low inherent membrane permeability and metabolic stability.
Drug screening methods for receptor targets typically encompass three approaches: (1) Identification of monoclonal antibodies (mAbs) via phage display, which has been employed for extracellular receptor complexes. (2) The high-throughput screening (HTS) of small molecules, a well-established strategy for targeting trans-membrane and intracellular receptor complexes, followed by optimization through medicinal chemistry. (3) Leveraging structural information or peptide display methods based on peptide modulators, such as SPOT, mRNA display, or phage display, which facilitate targeted drug development.
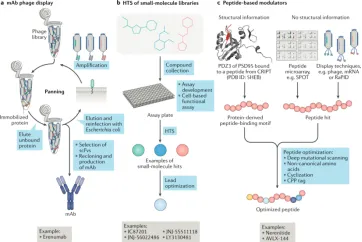
Figure 3. Strategies for Targeting Receptor Complexes [8]
(1) Antibodies
Antibodies are distinguished by their high affinity and selectivity, coupled with a prolonged duration of action. Compared to small molecular compounds, antibodies typically exhibit lower degradation rates. Moreover, antibodies can specifically target distinct conformations within receptor complexes, thus stabilizing either their active or inactive states. Recent advances in hybridoma technology and antibody phage display, especially when combined with HTS, have led to a substantial increase in the number of mAbs entering clinical trials.
The antibody phage display technique is extensively utilized for in vitro screening of fully humanized mAbs. This method involves the fusion of peptides to phage coat proteins, enabling the linkage of the phenotype (the displayed peptide) to the genotype (the DNA sequence within the same phage particle). The process entails cloning the entire antibody heavy and light chain genes. These genes are then expressed as either single-chain antibodies (scFv), single domain antibodies (VH or VL), or Fab antibodies. They are fused with the phage particle's structural protein (primarily the P3 protein) and displayed on the phage surface. By presenting antibodies on the phage surface and encapsulating the antibody genes, this technique effectively bridges the functional phenotype and genotype, allowing for the affinity-based selection of antibody genes through phenotypic screening, which capitalizes on the specific binding interaction between antigens and antibodies.

Figure 4. Illustrative Diagram of Phage Biopanning [9]
(2) Small Molecular Compounds
Small molecules are typically identified through HTS, necessitating high-quality, structurally diverse compound libraries, often comprising compounds from previous drug discovery projects focused on specific binding pockets. The development of meticulously designed and validated screening assays is essential for any HTS initiative. LGIC and GPCR screening assays generally involve cell-based functional assays, in contrast to mere binding assays.
Advancements in both in vitro and in vivo assays and screening techniques are hastening the identification of novel lead compounds targeting GPCR. These techniques encompass cell or membrane-based fluorescence screening, biosensors based on bioluminescence resonance energy transfer (BRET), and label-free approaches to drug discovery. For instance, in the case of GPCRs, the α (energy donor) and γ (energy acceptor) subunits of the G protein can be tagged with complementary fluorescent proteins, enabling BRET signal detection upon receptor activation. An effective approach to detect BRET signals is the use of fluorescent small molecule ligands, such as those containing BODIPY groups, in cells that express bioluminescent proteins fused to the C-terminal of GPCRs. BRET has been instrumental in identifying biased signaling and conformational changes or oligomerization in GPCRs. Chemiluminescence-based complementation assays, useful for β-arrestin recruitment, can be tailored for individual GPCRs to screen for agonists. Yeast-based fluorescent reporter systems, particularly those with optimized fluorescent proteins and engineered Gα subunits, have been effectively employed as functional biosensors for GPCR activity, notably in recent studies on serotonin receptors. Surface Plasmon Resonance (SPR) becomes applicable for compound screening when receptors are immobilized on gold surfaces.
(3) Peptides
Peptide drugs are a category of molecules created by linking multiple amino acids with peptide bonds, typically consisting of 10 to 100 amino acids. They are noted for their high efficiency, versatility, modifiability, and degradability, making them a significant interest. Development of peptide-based regulators can be facilitated using structural information or techniques like SPOT, mRNA display, and phage display. (1) Phage display involves cloning oligonucleotide fragments of synthesized random sequences into filamentous phages. Each phage then displays a specific peptide segment on its surface, and collectively, these phages form a library for screening binding peptides specific to target proteins. (2) Molecular-level in-situ screening coats target proteins onto 96-well plates, reducing interference from nonspecific factors due to the isolated nature of the targets. Solid-phase SPOT peptide synthesis is recognized for its efficiency in creating peptide libraries on membrane carriers, such as cellulose membranes, offering a rapid, high-throughput, and compatible approach for proteomics research. (3) Cellular-level screening compares cancer cells with normal cells to identify active peptides with high affinity for cancer cells, often targeting overexpressed or surface receptors. (4) Whole-body level screening involves injecting peptide phage libraries intravenously into animals, extracting and amplifying phages from tumor tissues. Through 3-5 rounds of biopanning, phages that bind specifically to tumor tissues are significantly enriched, leading to the identification of target peptides.
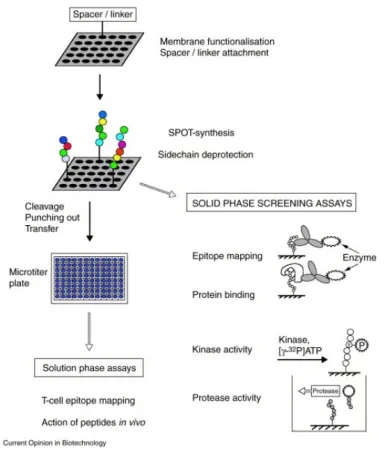
Figure 5. Formation of Peptide Arrays via SPOT Synthesis and Their Applications in Binding and Enzyme-Assisted Assays [10]
Analysis Workflow
1. Select Experimental Methodologies Based on Experimental Requirements
2. Preliminary Screening of Receptor Compound Libraries/Antibody or Peptide Screening via Phage Display
3. Secondary Screening and Validation of Small Molecules with Potential for Research
4. Analysis of Small Molecule Inhibition Mechanisms
5. Data Analysis
Service Advantages
1. High-credibility screening across a diverse range of receptor compound libraries.
2. Validation using multiple experimental methods, including radioisotope-labeled GPCR ligand binding tests or GTPγS binding tests; detection of downstream cAMP signaling following intracellular GPCR activation via homogenous time-resolved fluorescence; calcium flux detection induced by intracellular GPCR activation; detection of downstream inositol phosphate (IP3) following intracellular GPCR activation; protein phosphorylation assays; reporter gene-based assays, and others.
3. Mechanism analysis using high-credibility, high-precision mass spectrometry, among other techniques.
4. Comprehensive bioinformatics analysis.
Sample Results
1. Magnetic Bead-Based GPCR Phosphorylation Immunoassay for High-Throughput Ligand Analysis and GRK Inhibitor Screening [11]
The analysis of agonist-driven phosphorylation of GPCRs offers valuable insights into the activation states of receptors and the pharmacology of ligands. To date, the high-throughput assessment of GPCR phosphorylation has presented significant challenges. This study presents a validated magnetic bead-based immunoassay method for the quantitative evaluation of agonist-induced GPCR phosphorylation, which is fully compatible with multi-well cell culture plates. The assay includes the immunoprecipitation of affinity-tagged receptors using magnetic beads, followed by the detection of proteins with antibodies that are specific to and dependent on the phosphorylation state of GPCRs. The study involved treating five prototypical GPCRs (MOP, C5a1, D1, SST2, CB2) with a range of agonists and antagonists, generating concentration-response curves, and further developing the assay to establish a selective cellular GPCR kinase (GRK) inhibitor assay. This led to the rapid identification of selective inhibitors for GRK5/6 (LDC8988) and highly potent pan-GRK inhibitors (LDC9728).
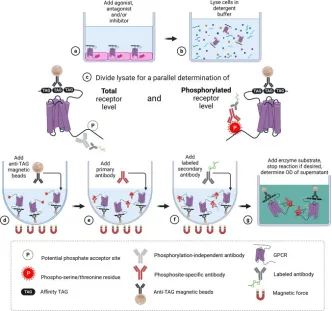
Figure 6. Experimental Procedure Diagram
2. Activation of Orphan Receptor GPR132 Induces Cell Differentiation in Acute Myeloid Leukemia [12]
The blockade of cell differentiation is a critical pathological feature of acute myeloid leukemia (AML). Research has demonstrated that the genetic activation of the orphan GPCR, GPR132, substantially triggers cell differentiation in AML, both in vitro and in vivo, indicating GPR132 as a potential trigger of myeloid differentiation. Investigating the therapeutic possibilities of GPR132 signaling, the study screened and validated 8-gingerol (8GL) as an agonist of GPR132. Remarkably, the activation of GPR132 by 8GL promoted differentiation in various human AML cell lines and reduced colony formation. Mechanistic studies revealed that 8GL treatment inhibits the activation of the mammalian target of rapamycin (mTOR), a regulator of AML cell differentiation blockade, via activating GPR132-Gs-PKA pathway. The combined application of 8GL and an mTOR inhibitor in vitro synergistically enhanced AML cell differentiation. Crucially, using 8GL, either independently or combined with an mTOR inhibitor, significantly hindered tumor growth and extended survival in mice, alongside improved cell differentiation.
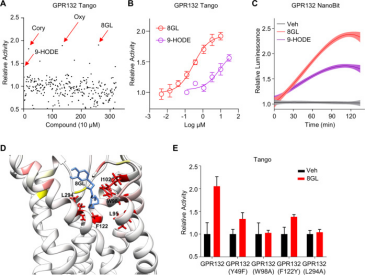
Figure 7. Screening and Validation of 8GL as a GPR132 Agonist
3. Biological Screening of a Unique Drug Library Targeting MRGPRX2 [13]
Allergic drug reactions, or drug allergies, represent a type of immune-mediated drug hypersensitivity reactions (DHR). GPCRs are frequent drug targets, mediating extracellular signals that initiate cellular responses. Recent findings emphasize the significant role of the GPCR MRGPRX2 in IgE-independent pseudoallergic DHRs, based on potential interactions between numerous peptidergic compounds and MRGPRX2. The study involved a primary HTS of 2,620 compounds from a library, targeting a mutation in MRGPRX3 N456S. Pharmacologically active compounds were discovered as MRGPRX2 agonists, their potency confirmed through calcium response assessments and degranulation assays. Employing the molecular tool Forge, the study also detailed the structure-activity relationships common to these identified active compounds.
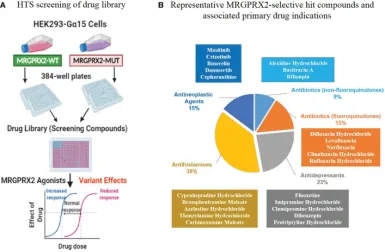
Figure 8. In vitro Biological High-Throughput Screening (HTS) of Drug Library to Identify MRGPRX2 Agonists
4. Identification of Effective Anticancer Drugs Through a Kinase and GPCR Polypharmacology Approach [14]
The complexity of pathogenic mechanisms and redundant signaling pathways often render the discovery of anticancer drugs targeting a single protein unsuccessful. Research has aimed to identify promising anticancer drug candidates by concurrently targeting casein kinase 1δ (CK1δ) and muscarinic acetylcholine receptor M3 (M3R). Through the structure-based virtual screening and de novo design, employing a modified protein-ligand binding potential function, a series of benzo[4,5]imidazo[1,2-a][1,3,5]triazin-2-amine (BITA) derivatives were identified as inhibitors of CK1δ and antagonists of M3R. These dual-function molecules exhibited biochemical potencies at nanomolar levels against CK1δ and low-micromolar levels against M3R. The interaction dynamics of the designed CK1δ-inhibitor and M3R-antagonist complexes notably involved the BITA segment, stabilized at the M3R orthosteric site and the CK1δ hinge region through forming three adjacent hydrogen bonds and hydrophobic contacts.
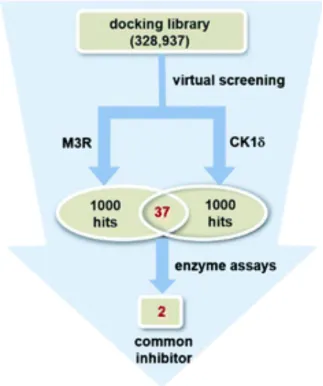
Figure 9. Discovery of Dual M3R-Antagonists and CK1δ-Inhibitors through Parallel Virtual and Biochemical Screening
Sample Submission Requirements
1. Please provide pre-purified samples wherever possible.
2. Efforts should be made to minimize impurity contamination.
Services at MtoZ Biolabs
1. Detailed experimental procedures.
2. Specifications of relevant instruments.
3. Raw experimental data.
4. Comprehensive reports on the screening of sample types such as small molecule inhibitors, including rankings of inhibitory effects and information on inhibitory IC50 values.
Applications
1. The Inhibition/Activation of GPCR Receptors for Clinical Treatment
Current AD therapies, for instance, try to ameliorate cholinergic transmission deficits by inhibiting acetylcholinesterase to prevent acetylcholine degradation, yet these therapies have limited clinical effectiveness. An alternative strategy involves directly activating cholinergic receptors associated with learning and memory. The M1 muscarinic acetylcholine receptor (mAChR) is an optimal target, although its application is limited by adverse effects. There are ongoing efforts to define the drug properties necessary for mAChR agonists that are well tolerated. This involves a multi-step translation process, including the utilization of atomic structures, cell/tissue assays, preclinical species evaluation, clinical safety trials, and ultimately verifying activity in human memory centers. This approach has led to the rational design of properties like selectivity and partial agonism, culminating in the development of HTL9936, a promising drug candidate for addressing memory loss in AD.
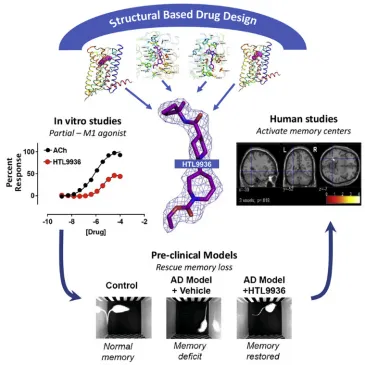
Figure 10. Synthesis and Clinical Application of Partial Agonists for the mAChR[15]
FAQ
Q1: Besides experimental screening, what other methods can be utilized to refine the selection of candidate compounds?
Receptor-based virtual screening methods offer an initial screening approach. Known as structure-based virtual screening, this technique requires the three-dimensional structure of the target. It encompasses a series of computational steps: preparing the target and the molecular library, conducting docking and post-docking analysis, and establishing a priority order for compounds to be tested experimentally. The method entails docking each ligand molecule into the target's binding site, predicting the binding mode for each ligand, and assessing the quality of how well the ligand fits within the binding site of the target.
References
[1] Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci. 2019 Aug;40(8):592-604. doi: 10.1016/j.tips.2019.06.004. Epub 2019 Jul 15. Erratum in: Trends Pharmacol Sci. 2019 Oct;40(10):801. PMID: 31320117.
[2] Pinzi L, Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int J Mol Sci. 2019 Sep 4;20(18):4331. doi: 10.3390/ijms20184331. PMID: 31487867; PMCID: PMC6769923.
[3] Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020 Mar 28;395(10229):1078-1088. doi: 10.1016/S0140-6736(20)30164-1. PMID: 32222192.
[4] Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017 Jul 27;170(3):414-427. doi: 10.1016/j.cell.2017.07.009. PMID: 28753422; PMCID: PMC5560499.
[5] Choi S, Choi KY. Screening-based approaches to identify small molecules that inhibit protein-protein interactions. Expert Opin Drug Discov. 2017 Mar;12(3):293-303. doi: 10.1080/17460441.2017.1280456. Epub 2017 Jan 20. PMID: 28067063.
[6] Dunn HA, Orlandi C, Martemyanov KA. Beyond the Ligand: Extracellular and Transcellular G Protein-Coupled Receptor Complexes in Physiology and Pharmacology. Pharmacol Rev. 2019 Oct;71(4):503-519. doi: 10.1124/pr.119.018044. PMID: 31515243; PMCID: PMC6742926.
[7] Yang D, Zhou Q, Labroska V, Qin S, Darbalaei S, Wu Y, Yuliantie E, Xie L, Tao H, Cheng J, Liu Q, Zhao S, Shui W, Jiang Y, Wang MW. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct Target Ther. 2021 Jan 8;6(1):7. doi: 10.1038/s41392-020-00435-w. PMID: 33414387; PMCID: PMC7790836.
[8] Rosenbaum MI, Clemmensen LS, Bredt DS, Bettler B, Strømgaard K. Targeting receptor complexes: a new dimension in drug discovery. Nat Rev Drug Discov. 2020 Dec;19(12):884-901. doi: 10.1038/s41573-020-0086-4. Epub 2020 Nov 11. PMID: 33177699.
[9] Alfaleh MA, Alsaab HO, Mahmoud AB, Alkayyal AA, Jones ML, Mahler SM, Hashem AM. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front Immunol. 2020 Aug 28;11:1986. doi: 10.3389/fimmu.2020.01986. PMID: 32983137; PMCID: PMC7485114.
[10] Reineke U, Volkmer-Engert R, Schneider-Mergener J. Applications of peptide arrays prepared by the SPOT-technology. Curr Opin Biotechnol. 2001 Feb;12(1):59-64. doi: 10.1016/s0958-1669(00)00178-6. PMID: 11167074.
[11] Kaufmann J, Blum NK, Nagel F, Schuler A, Drube J, Degenhart C, Engel J, Eickhoff JE, Dasgupta P, Fritzwanker S, Guastadisegni M, Schulte C, Miess-Tanneberg E, Maric HM, Spetea M, Kliewer A, Baumann M, Klebl B, Reinscheid RK, Hoffmann C, Schulz S. A bead-based GPCR phosphorylation immunoassay for high-throughput ligand profiling and GRK inhibitor screening. Commun Biol. 2022 Nov 9;5(1):1206. doi: 10.1038/s42003-022-04135-9. PMID: 36352263; PMCID: PMC9646841.
[12] Yi C, He J, Huang D, Zhao Y, Zhang C, Ye X, Huang Y, Nussinov R, Zheng J, Liu M, Lu W. Activation of orphan receptor GPR132 induces cell differentiation in acute myeloid leukemia. Cell Death Dis. 2022 Nov 27;13(11):1004. doi: 10.1038/s41419-022-05434-z. PMID: 36437247; PMCID: PMC9701798.
[13] Yang F, Limjunyawong N, Peng Q, Schroeder JT, Saini S, MacGlashan D Jr, Dong X, Gao L. Biological screening of a unique drug library targeting MRGPRX2. Front Immunol. 2022 Oct 21;13:997389. doi: 10.3389/fimmu.2022.997389. PMID: 36341461; PMCID: PMC9635925.
[14] Park H, Jung HY, Mah S, Kim K, Hong S. Kinase and GPCR polypharmacological approach for the identification of efficient anticancer medicines. Org Biomol Chem. 2020 Oct 28;18(41):8402-8413. doi: 10.1039/d0ob01917h. PMID: 33112339.
[15] Brown AJH, Bradley SJ, Marshall FH, Brown GA, Bennett KA, Brown J, Cansfield JE, Cross DM, de Graaf C, Hudson BD, Dwomoh L, Dias JM, Errey JC, Hurrell E, Liptrot J, Mattedi G, Molloy C, Nathan PJ, Okrasa K, Osborne G, Patel JC, Pickworth M, Robertson N, Shahabi S, Bundgaard C, Phillips K, Broad LM, Goonawardena AV, Morairty SR, Browning M, Perini F, Dawson GR, Deakin JFW, Smith RT, Sexton PM, Warneck J, Vinson M, Tasker T, Tehan BG, Teobald B, Christopoulos A, Langmead CJ, Jazayeri A, Cooke RM, Rucktooa P, Congreve MS, Weir M, Tobin AB. From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer's disease. Cell. 2021 Nov 24;184(24):5886-5901.e22. doi: 10.1016/j.cell.2021.11.001. PMID: 34822784.
How to order?

