





Sample Preparation Guide for Phosphorylated Protein Mass Spectrometry Analysis
- Use lysis buffers containing phosphatase inhibitors (e.g., NaF, Na3VO4, β-glycerophosphate).
- Urea-based lysis is recommended, or SDS-based lysis followed by on-column desalting to improve protein recovery.
- Perform procedures rapidly and at low temperature whenever possible to minimize protease and phosphatase activity.
- When feasible, use the BCA assay for protein quantification to reduce potential interference from Tris in colorimetric reactions.
- Trypsin digestion is recommended, optionally combined with a Trypsin + Lys-C dual-enzyme strategy to improve cleavage efficiency.
- Control digestion time (12–16 h) and temperature (37°C) to minimize excessive degradation or enzyme autodigestion.
- Desalting using C18 solid-phase extraction (SPE) cartridges
- SpeedVac drying/concentration prior to injection
- Reconstitution in solutions containing 0.1% FA to facilitate chromatographic separation
Protein phosphorylation is among the most prevalent and biologically important post-translational modifications (Post-Translational Modification, PTM) and is broadly involved in cellular signal transduction, cell-cycle control, metabolic regulation, and other essential biological processes. Systematic identification and quantitative analysis of phosphorylated proteins have become an active area of modern life science research, with significant applications in oncology, neurodegenerative disorders, and autoimmune diseases. Advances in mass spectrometry (Mass Spectrometry, MS) have enabled large-scale phosphoproteomic studies. However, relative to whole-proteome profiling, sample preparation for phosphorylated proteins is more complex and requires tighter experimental control. Appropriate sample handling strategies directly determine the depth and quality of downstream MS data.
Challenges and Strategies in Phosphorylated Protein Analysis
1. Low Abundance and High Dynamic Range: Intrinsic Challenges
Phosphorylation sites are often low in abundance, transient, and reversible. In cells, phosphopeptides typically account for only ~1%–2% of the total peptide population; therefore, direct MS analysis frequently yields suboptimal coverage. The central strategy to address this limitation is phosphopeptide enrichment.
2. Sample Preparation Is a Prerequisite for Robust Mass Spectrometry Outcomes
Inappropriate lysis buffers, incomplete enzymatic digestion, or protein degradation/dephosphorylation can compromise reproducibility, reduce the number of confidently identified sites, and introduce substantial quantitative bias. A standardized sample preparation workflow is essential for experimental stability and reproducibility.
Five-Step Workflow for Phosphorylated Protein Sample Preparation
Step 1: Sample Lysis and Protein Extraction
(1) Objective: to maximize recovery of target proteins while preserving phosphorylation status as much as possible.
(2) Key elements:
Step 2: Protein Quantification and Enzymatic Digestion
(1) Objective: to accurately determine protein concentration and ensure efficient digestion.
(2) Notes:
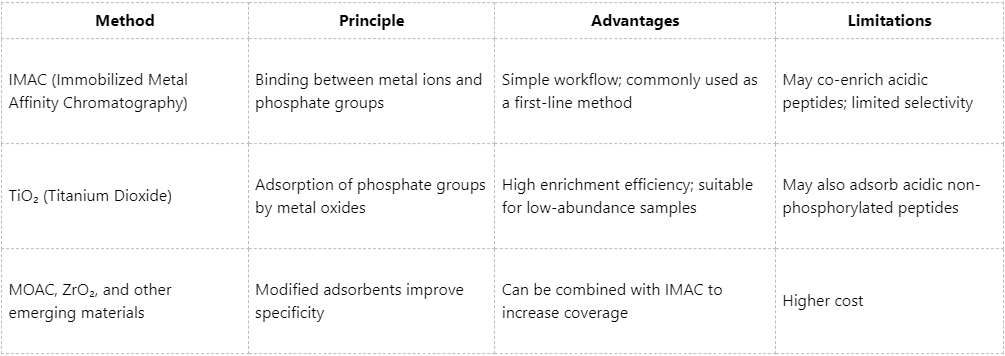
Step 3: Phosphopeptide Enrichment
(1) Objective: to selectively enrich phosphopeptides from complex peptide mixtures.
(2) Comparison of mainstream enrichment methods:
Step 4: Sample Cleanup and Pre-Injection Processing
(1) Objective: to remove salts, buffers, and impurities and ensure compatibility with LC-MS requirements.
(2) Common approaches:
Step 5: Mass Spectrometry Acquisition and Data Analysis
The choice of MS platform, instrument parameter settings, database searching, and statistical processing also influence the final data depth and confidence.
Additional Recommendations to Improve Phosphoproteomics Data Quality
1. Rational Experimental Design and Replication
(1) It is recommended to include ≥3 biological replicates per group to improve statistical confidence.
(2) For quantitative experiments (e.g., TMT labeling), batch effects should be carefully controlled.
2. Emphasize Site-Level Quantification Rather Than Protein-Level Quantification
Because phosphorylation is highly site-specific, biological interpretation should be grounded in site-level quantification.
3. Leverage Bioinformatic Mining to Increase Data Value
(1) Motif analysis, pathway enrichment, and kinase prediction can be used to infer potential regulatory networks.
(2) Recommended resources include: PhosphoSitePlus, KEGG, KinaseSubstrateDB.
Mass spectrometry-based analysis of phosphorylated proteins is a key approach for elucidating cellular signaling networks, and high-quality sample preparation provides the foundation for this process. If you encounter technical challenges during implementation or wish to apply phosphoproteomics to disease research or drug development, please feel free to contact MtoZ Biolabs. We will tailor solutions to your specific needs.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

