





Recombinant Protein Drug Analysis Service
Recombinant protein drugs are produced using recombinant DNA or RNA technologies. This involves the integration of target protein genes into recombinant vectors, which are then introduced into host cells to synthesize specific recombinant proteins. These drugs are primarily designed to replace or supplement proteins that are deficient due to genetic disorders or diseases.
The primary production systems for these drugs include prokaryotic systems (e.g., E. coli), mammalian systems (e.g., CHO and HEK293 cells), eukaryotic systems (e.g., yeast), and insect cell systems. Additionally, transgenic animals and plants can also be used to produce recombinant proteins.
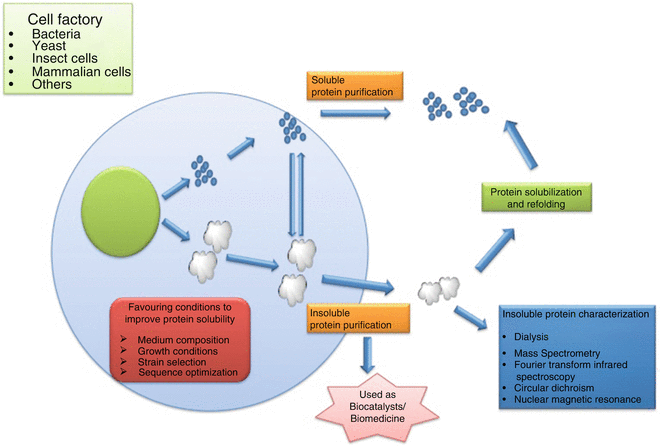
Figure 1. Production of Recombinant Proteins [1]
A wide variety of recombinant protein drugs are available, including peptide hormones, cytokines, and recombinant enzymes. For example, recombinant human insulin is an important drug in the treatment of diabetes, while recombinant human growth hormone is used in the treatment of growth hormone deficiency. In addition, recombinant protein drugs include recombinant interferon and erythropoietin, which play an important role in the treatment of hepatitis, tumors, and other diseases.
Advantages and Limitations of Recombinant Proteins as Therapeutic Drugs
1. Advantages
(1) High Specificity and Efficacy: Recombinant protein drugs exhibit specific biological activities, targeting distinct biomarkers or pathological processes efficiently.
(2) Broad Therapeutic Range: They are applicable in treating a diverse array of conditions, including cancer, genetic disorders, immune-related diseases, and chronic illnesses.
(3) Low Toxicity and Side Effects: These drugs generally offer lower toxicity and fewer side effects compared to conventional small molecule drugs, as they are based on naturally occurring human proteins.
(4) Biocompatibility: Recombinant proteins are well-tolerated by the human body due to their similarity to natural human proteins.
(5) Long-Lasting Effects: Technologies like PEGylation and protein fusion extend the half-life of these drugs in the body, enhancing patient compliance by reducing the dosing frequency.
2. Limitations
(1) High Production Costs: Their production is complex and requires advanced biotechnological platforms and stringent production environments, leading to higher costs.
(2) Stability Concerns: These drugs may face stability issues during storage or transport, necessitating specific conditions to retain their activity.
(3) Potential Immunogenicity: Despite their design to mimic natural proteins, recombinant protein drugs can still elicit immune responses, possibly reducing their effectiveness or causing adverse effects.
(4) Restricted Administration Routes: Most are administered via injection, which may limit patient convenience and adherence.
(5) Extended Development Timelines: Their development involves lengthy clinical trials and regulatory processes.
Strategies to Overcome Limitations Associated with Recombinant Proteins
1. Enhancing Expression
Optimization of expression vectors, selection of suitable host cells, and improvement of culture conditions can substantially boost production yields. For example, utilizing strong promoters and effective signal peptides enhances protein expression and secretion.
2. Improving Stability and Folding
Employing molecular chaperones or stabilizers, and fusion techniques to combine target proteins with stabilizers, improves their solubility and stability. Protein engineering, such as humanization, can also reduce immunogenicity and enhance stability.
3. Reducing Immunogenicity
Modifying antibody constant regions or using fully human antibody libraries can diminish the immunogenic potential of antibody drugs. Predicting T-cell epitopes within antibody sequences is another effective strategy.
4. Optimizing Delivery Systems
Developing non-injection formulations, such as transdermal patches, mucosal absorptions, inhalable sprays, and oral dosages, improves patient compliance and minimizes side effects associated with injections.
5. Advancing Modification Techniques
Genetic and chemical modifications, such as PEGylation, alter drug pharmacokinetics and reduce immunogenicity. Exploring new materials like helical polyamino acids (PEPylation) as alternatives to PEG represents a promising direction.
6. Enhancing Purity and Quality Control
Improvements in purification processes and quality control ensure high purity and consistency of recombinant protein drugs.
7. Focusing on Stability and Formulation Development
Detailed stability studies and the development of solutions for high-concentration protein formulations address challenges in ensuring long-term stability and effectiveness of these drugs.
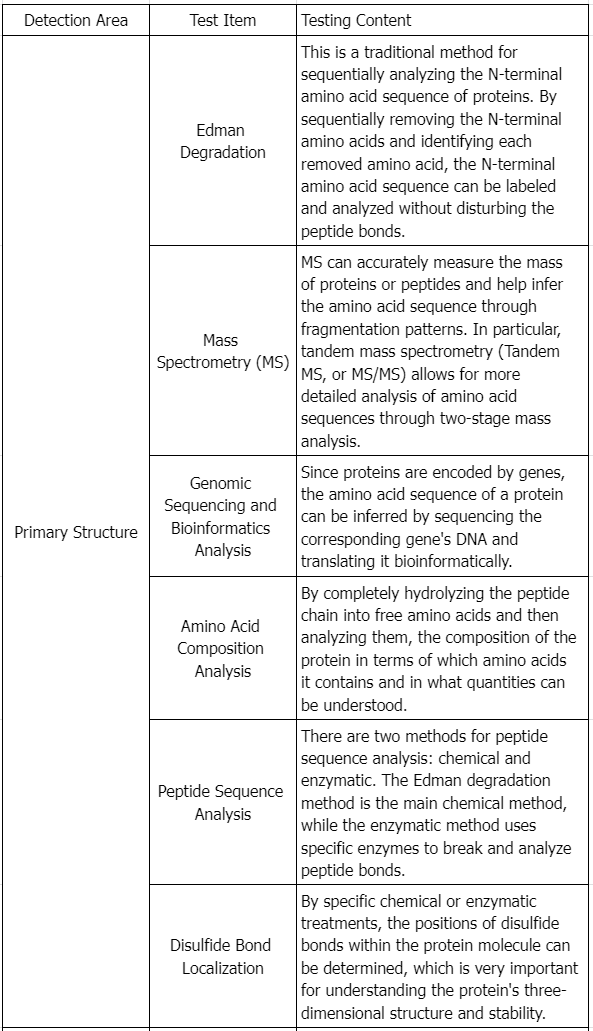
Testing Content Required by Policies and Regulations
Draft Guidance of Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations
To assess if a proposed product and its reference product are highly similar, extensive and robust comparative structural and functional studies are required. These studies should include biological assays, binding assays, and enzymatic kinetics. The similarity assessment relies on the latest analytical capabilities, for example, the molecular weight of the protein, complexity of the protein (higher order structure and post-translational modifications), degree of heterogeneity, functional properties, impurity profiles, and degradation profiles denoting stability. The applicant must clearly describe the methods employed in these assessments and address their limitations. These studies aim to establish essential quality attributes defining the product's identity, quantity, safety, purity, and potency. The product-related impurities and product-related substances should be identified, characterized as appropriate, quantified, and compared using multiple lots of the proposed product and multiple lots of the reference product, to the extent feasible and relevant, as part of an assessment of the potential impact on the safety, purity, and potency of the product.
An extensive analytical characterization may reveal differences between the proposed product and reference product, especially when analytical techniques are used to identify qualitative or quantitative differences in product attributes. Developing orthogonal quantitative methods to distinctly differentiate any product attribute variations is crucial. Following the functional and physicochemical properties assessment (which includes higher order structures, post-translational modifications, impurities, and degradation profiles), the applicant should choose targeted approaches for further animal and/or clinical studies to support biosimilarity claims. Integrating a vast array of product attributes using high sensitivity orthogonal methods alongside fingerprint-like analytical algorithms proves beneficial in comparing quality attribute differences. Enhancing production's scientific basis, as discussed in ICH Q8 (R2), facilitates better quality matching during manufacturing. This strategy quantifies the overall similarity between the molecules, offering selective and targeted approaches for future animal and/or clinical studies.
When evaluating high similarity, manufacturers should consider multiple factors, including:
1. Expression Systems
Therapeutic proteins may originate from microbial cells (prokaryotic or eukaryotic), cell lines (e.g., mammalian, avian, insect, plant), or tissues derived from animals or plants. The expression construct of the proposed product should encode the same primary amino acid sequence as its reference product. Minor modifications, like N- or C-terminal truncations (e.g., C-terminal lysine heterogeneity in monoclonal antibodies), expected not to affect performance, should be adjusted and explained by the applicant. The chosen expression systems' differences, affecting the process and related substances, impurities, and potential contaminants (including potential adventitious agents), need careful consideration. The impact of the expression system on translational and post-translational modifications could introduce additional uncertainties in achieving high similarity with the reference product. Minimizing these differences enhances the likelihood of producing highly similar protein products, evaluated on a case-by-case basis.
2. Manufacturing Process
A comprehensive understanding of all stages in the manufacturing process for the proposed is essential. Specific information from characterization tests, process controls, and specifications should relate directly to the proposed product and its manufacturing process. Enhancing drug development methods, quality risk management, and effective quality systems promotes high-quality production. For 351(a) BLAs, drug intermediates, or products not applicable for 351(k) applications as license holders need insights into biologic production processes and control. If the applicant opts not to produce licensed products, alternative contract manufacturing arrangements might be considered.
A sponsor considering manufacturing changes after completing the initial comparative analytical assessment or after completing clinical studies intended to support a 351(k) application will need to demonstrate comparability between the pre- and post-change proposed product and may need to conduct additional studies. The nature and extent of these changes dictate the additional similarity studies required. If process materials differ from those in clinical trials, analytical similarity studies should include a sufficient batch number of the proposed biosimilar.
3. Physicochemical Properties Assessment
Assessing the physicochemical properties of the proposed product and reference product involves considering all pertinent protein characteristics (e.g., primary, secondary, tertiary, and quaternary structures, post-translational modifications, and functional activities). The goal is to detect any differences in quality attributes between the two products maximally.
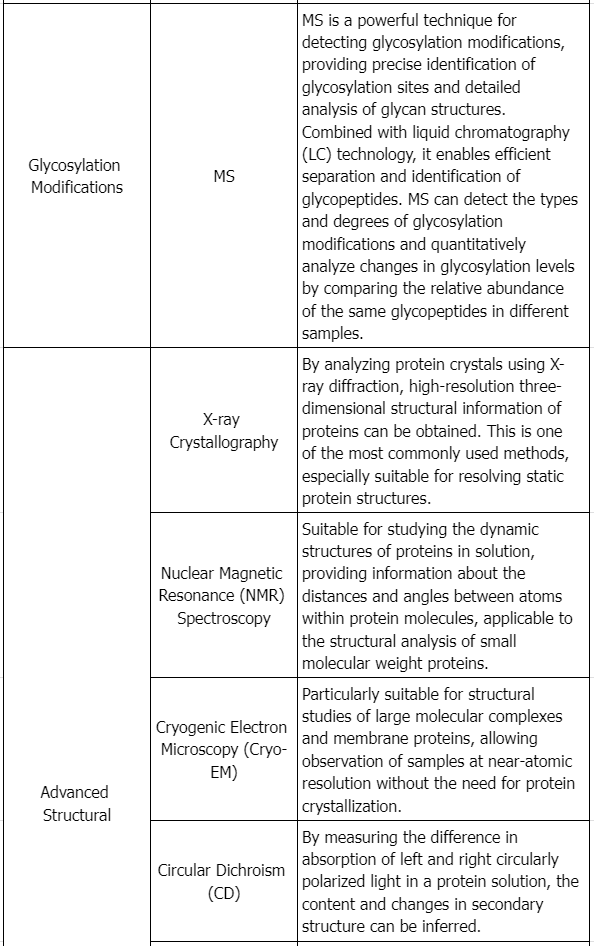
In guiding characterization studies, applicants should focus on the product concepts discussed in ICH Q6B, recognizing the importance of understanding variations such as different subtypes, glycosylation levels, and changes from post-translational modifications.
Specific analytical methodologies for assessing proteins' physicochemical properties are outlined in scientific literature, regulatory guidelines, and pharmacopeial compendia, which provide essential information for selecting appropriate analytical approaches critical to product performance based on the characterized protein's nature and the structural and heterogeneity knowledge of the reference and proposed products.
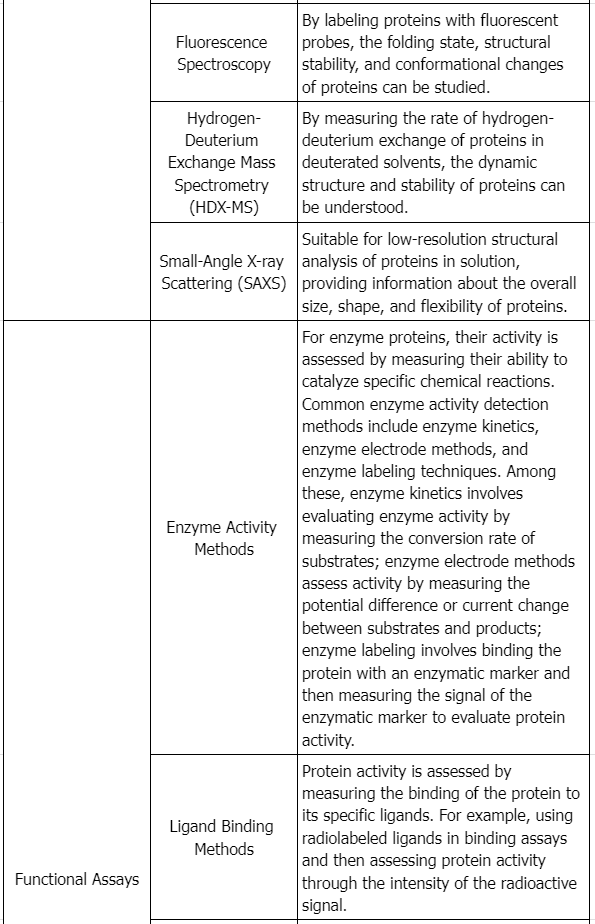
4. Functional Activity
Functional assays play multiple roles in protein product characterization, supplementing physicochemical analyses as qualitative measures of functionality.
While physicochemical analysis may not always confirm the integrity of higher order structures due to the protein's complexity and the limits of current analytical techniques, biological activity often indicates structural integrity. If the action mechanism of the reference product is known or can be reasonably inferred, functional analyses should, as closely as possible, reflect these mechanisms. Conducting a variety of functional analyses is crucial in assessing analytical similarity, with evaluations of functional activity providing insights into production consistency, purity, potency, and stability.
When a reference product exhibits multiple functional activities, the applicant must conduct a range of tests assessing these activities' scope. For example, for proteins with various functional domains and receptor-mediated activities, it is essential to evaluate both activity types. Functional activities can be measured across multiple parameters, such as enzymatic kinetics or interactions with blood clotting factors, to assess each product parameter comprehensively.
Applicants must recognize potential limitations of some functional assays, like high variability, which might obscure significant differences between the proposed product and the reference product. Tests with less variability that are sensitive to functional activity changes are recommended. Additionally, in vitro biological activity may not entirely represent a protein's clinical activity, as these tests often do not account for bioavailability, influencing pharmacodynamics and clinical performance. Given the subtle differences affecting distribution and post-translational modifications, these limitations must be considered when evaluating the data quality or obtaining additional information supporting biosimilarity, addressing any residual uncertainties. Ultimately, functional assays are indispensable in assessing neutralizing antibody methods in both preclinical and clinical research phases.
5. Receptor Binding and Immunochemical Properties
When binding or immunochemical properties are part of a protein product's activity, analytical tests should be conducted to describe the attributes of the proposed product in these specific properties (for instance, if receptor binding is an inherent protein function, this property should be measured and used in comparative studies) (see ICH Q6B to get more information). Various methods such as surface plasmon resonance, microcalorimetry, or classical Scatchard analysis can provide information about the kinetics and thermodynamics of binding. Such information relates to the functional activity and characteristics of the proposed product's higher order structure.
6. Impurities
Applicants should characterize, identify, and quantify the impurities of the proposed product and reference product as feasibly as possible. Differences in the production process impurities of the proposed product and reference product should be assessed using risk-based methods. If comparative physicochemical analysis reveals similar levels of relevant impurities in both products, pharmacological/toxicological studies to characterize the potential biological effects of specific impurities may be unnecessary. However, if the production process used for the proposed product introduces different impurities, or if the levels are higher than those in the reference product, additional pharmacological/toxicological or other studies may be necessary. As discussed in the ICH guidance for industry S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals, “it is preferable to rely on purification processes to remove impurities . . . rather than to establish a preclinical testing program for their qualification.”
Evaluate process-related impurities generated by cell substrates (e.g., host cell DNA and host cell proteins), cell culture components (e.g., antibiotics and culture media components), and downstream processing steps (e.g., reagents, solvent residues, leachables, endotoxins, and bioburden). Process-related impurities in the proposed product are expected not to match those observed in the reference product. However, impurities related to the manufacturing process of both the proposed product and the reference product should be evaluated concurrently. The different impurity profiles' impact on safety should be addressed and supported by appropriate data. A specific approach will be used to evaluate the differences between the proposed product and the U.S. marketed reference product and consider the applicant's assessment of the potential impact of these differences on the biosimilar. In all cases, the chosen analytical methods should be capable of adequately detecting, identifying, and accurately quantifying biologically significant levels of impurities (refer to ICH industry guideline Q2B "Validation of Analytical Procedures: Methodology"). Additionally, the results of immunological method detections depend on the host cell protein assay reagents and cell matrix used. Product cell substrates and orthogonal methods should be used to validate this test, ensuring accuracy and sensitivity. This should be performed based on the relatedness and feasibility of the two products.
Regarding exogenous or endogenous viral contamination of the proposed product and the safety of any biologic, it should be ensured by screening critical raw materials and confirming the removal of robust viruses and achieving inactivation during the production process (see ICH industry guideline Q5A "Viral Safety Evaluation of Biotechnology Products Produced from Human or Animal Cell Lines").
7. Stability
As part of an appropriate physicochemical and functional comparison of the stability profile of the proposed product with that of the reference product, accelerated and stress stability studies, as well as forced degradation studies, should be used to establish degradation profiles and to provide a direct stability comparison of the proposed product with the reference product. These comparative studies should be conducted under multiple stress conditions (e.g., high temperatures, freeze thaw, light exposure, and agitation), which may cause incremental degradation of the product over specified time periods. These studies can reveal product differences and identify additional control conditions used during production and storage (see ICH industry guidelines Q5C "Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products and Q1A (R2) Stability Testing of New Drug Substances and Products"). Sufficient time, real-condition stability data should be provided for the proposed product to support the suggested shelf life.
Recombinant Protein Drug Analysis Solutions
Service Advantages
1. Professional and Robust Services Covering a Wide Range of Recombinant Protein Drug Testing
2. High Cost-Effectiveness, Short Testing Cycles, and Reliable, Comprehensive Test Reports
3. Equipped with a Variety of High-Resolution MS and other Major Equipment
Sample Results
1. Infrared Spectroscopy for Recombinant Protein Quality Assessment: Characterization of Ca²⁺-ATPase Protein Aggregation-Related Bands [2]
Infrared spectroscopy was utilized to characterize recombinant sarcoplasmic reticulum Ca2+-ATPase (SERCA1a). In the amide I region, its spectrum differed below 1650 cm−1 compared to the spectrum of Ca2+-ATPase prepared from rabbit fast twitch muscle. The band at 1642 cm−1 in the recombinant protein's spectrum was reduced, while the band at 1631 cm−1 was more prominent. By comparison of amide I band areas with the known secondary structure content of the protein, we assigned the 1642 cm−1 band to β-sheet structure. Further investigations indicated a decrease in the 1642 cm−1 band after room temperature storage and repeated water washes of the protein membrane, while the 1631 cm−1 band increased. Protein aggregates formed after solubilizing rabbit muscle enzymes showed a prominent band at 1631 cm−1, resembling the spectrum of membrane bound ATPase. The presence of the 1631 cm−1 band, similar to other protein inclusions, suggested protein aggregation, serving as a quality marker for optimizing recombinant protein production. The study concluded that the recombinant production of SERCA1a, storage at room temperature, repeated washing and aggregation after solubilisation all modify existing β-sheets in the cytosolic domains so that they become similar to those found in inclusion bodies of other proteins.
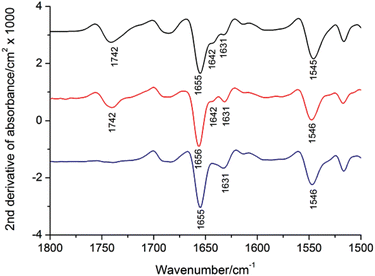
Figure 2. Second Derivative Absorption Spectra of Rabbit SERCA1a before and after Detergent Solubilization
Black: membrane bound SERCA1a; Red: solubilized SERCA1a in 1 mg/mL DDM; Blue: insolubilised pellet.
2. Quality Assessment of Recombinant Proteins Produced in Plant [3]
The acceptance of plant-based recombinant protein expression technologies by the pharmaceutical and other commercial markets highlights the safer, more cost-effective, and less capital-intensive alternatives to traditional microbial fermentation or animal cell culture bioreactors. Plants are capable of expressing biologically active proteins from various species, including humans and animals. As plant-produced proteins move to commercial markets, developing methods to evaluate their quality and performance becomes essential for supporting QA/QC requirements. Compliance with the ICH Q6B guidelines is crucial for biologics' process validation and acceptance criteria in the pharmaceutical industry. Detailed product specifications also need to be developed and validated for plant proteins used in bioenergy, food, chemical synthesis, or as research reagents.
Therefore, methods have been developed to assess important qualitative and quantitative parameters of the product and the manufacturing methods used in plant production systems. This chapter outlines procedures for validating product characteristics, including quality analysis, antibody cross-reactivity, N-terminal sequencing, and bioactivity. It introduces standard methods for routine assessment of yield, recovery, and purity, developed specifically for synthesizing and recovering the avian cytokine, chicken interleukin-12 (ChIL-12) produced in tobacco leaves. The ChIL-12 protein, used here as a model with a C-terminal histidine epitope (HIS-tag), may be directly applicable to other HIS-tagged proteins produced in plants. The overall strategies outlined for ChIL-12(HIS) should serve as a standard procedural basis for assessing the quality of other plant-based protein products and production systems.
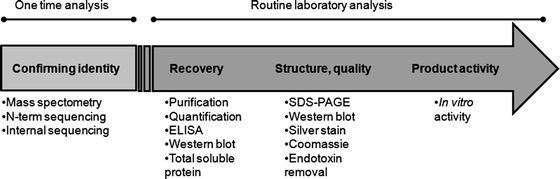
Figure 3. Workflow for Quality Assessment of Recombinant Protein Products
References
[1] Ferrer-Miralles N, Saccardo P, Corchero JL, Xu Z, García-Fruitós E. General introduction: recombinant protein production and purification of insoluble proteins. Methods Mol Biol. 2015;1258:1-24. doi:10.1007/978-1-4939-2205-5_1.
[2] Li C, Kumar S, Montigny C, le Maire M, Barth A. Quality assessment of recombinant proteins by infrared spectroscopy. Characterisation of a protein aggregation related band of the Ca²⁺-ATPase. Analyst. 2014;139(17):4231-4240. doi:10.1039/c4an00483c.
[3] Medrano G, Dolan MC, Condori J, Radin DN, Cramer CL. Quality assessment of recombinant proteins produced in plants. Methods Mol Biol. 2012;824:535-564. doi:10.1007/978-1-61779-433-9_29.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Host Cell Protein Assay Service
Protein Characterization Service
Protein Structure Identification Service
How to order?

