





Bottom-Up Proteomics Data Analysis Pipeline: From Sample Prep to Pathway Interpretation
- Digestion QC precedes downstream claims.
- DDA for discovery depth; DIA for scalable quantification.
- Document search, FDR, and inference settings.
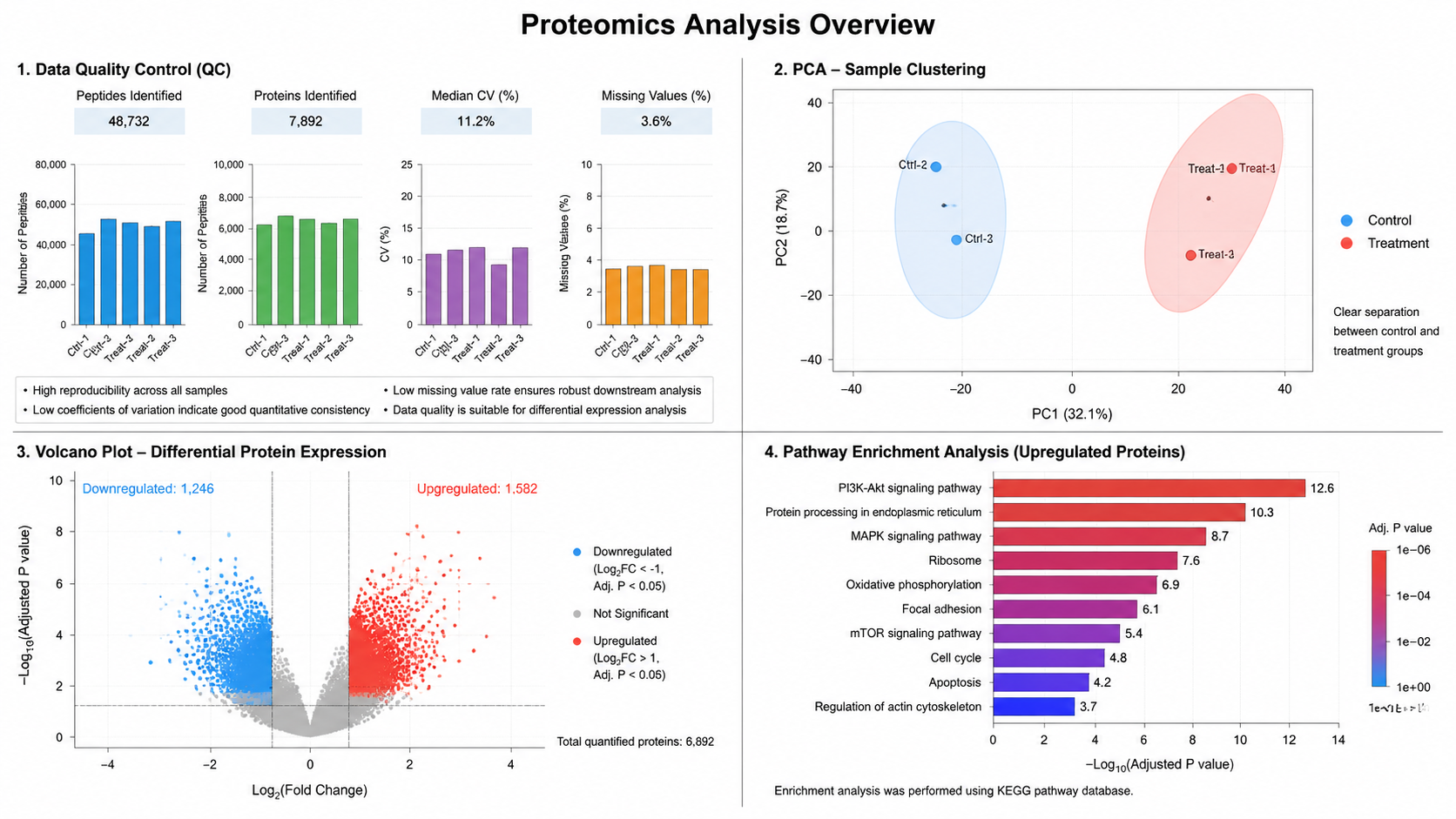
- PCA and normalization catch batch effects.
- Enrichment translates lists into hypotheses.
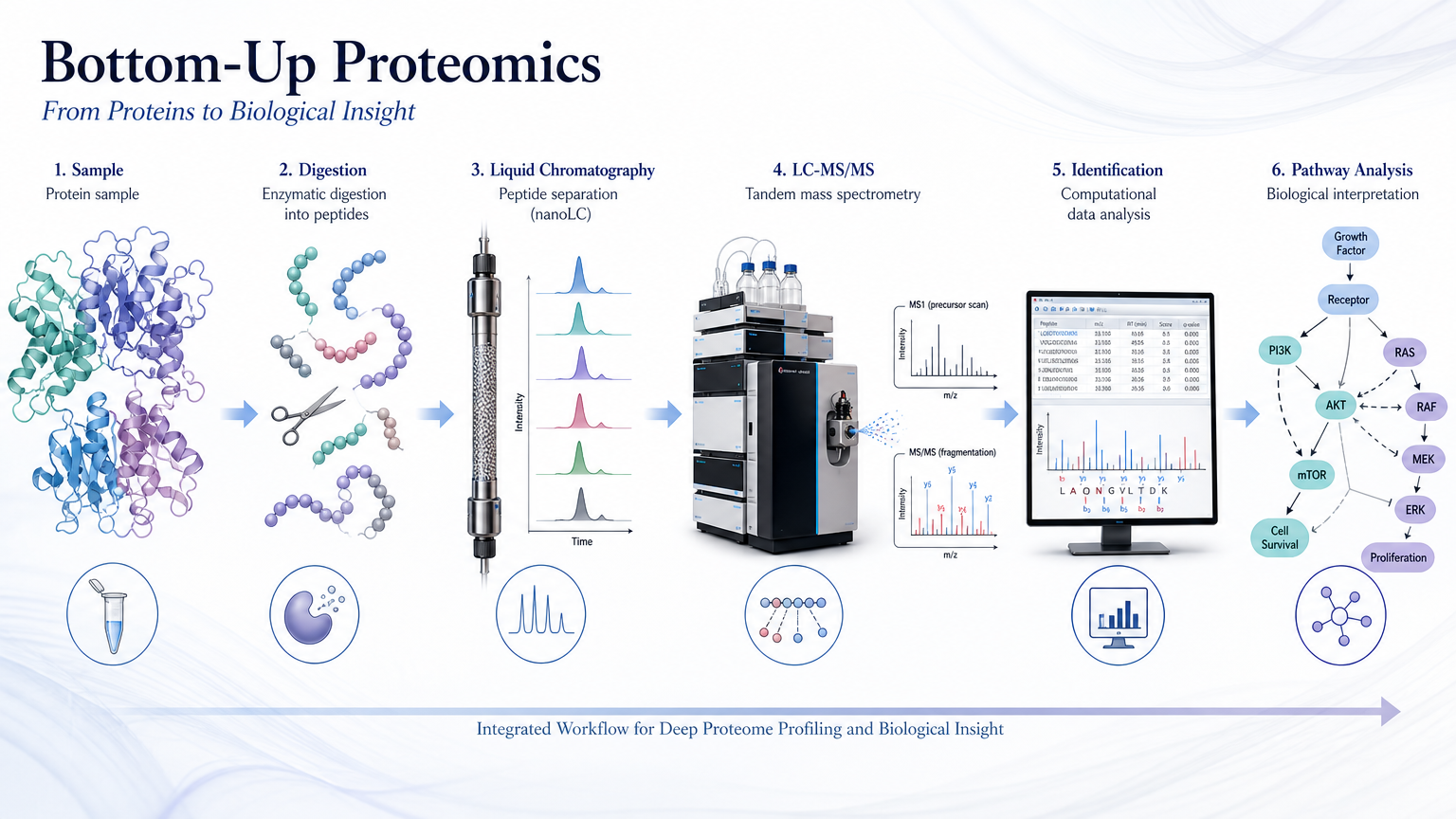
Bottom-up proteomics digests proteins to peptides, identifies them by LC-MS/MS, and infers protein abundance. A standardized pipeline keeps large studies reproducible from extraction through pathway interpretation.
Key Takeaways
Related Services
Bioinformatics Customized Service
Proteomics Analysis Services, Biopharmaceutical Characterization Services, Bioinformatics Services
Sample Preparation and LC-MS/MS
Extract and quantify protein; digest with trypsin; separate peptides by nanoLC; acquire DDA or DIA on high-resolution MS.
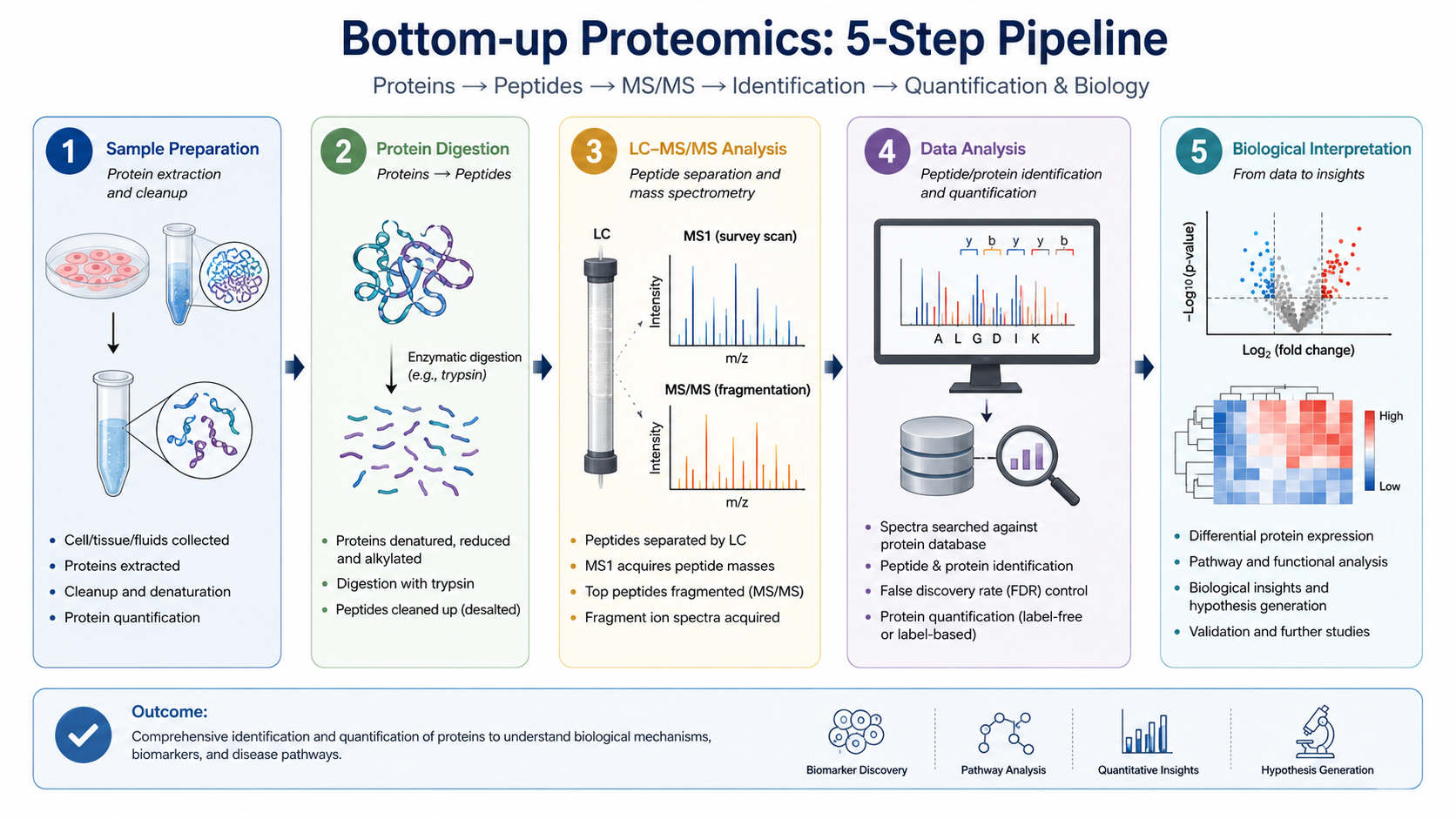
Raw Data Processing
Convert raw files; search databases at ~1% FDR; infer proteins; quantify label-free, TMT, or DIA-based.
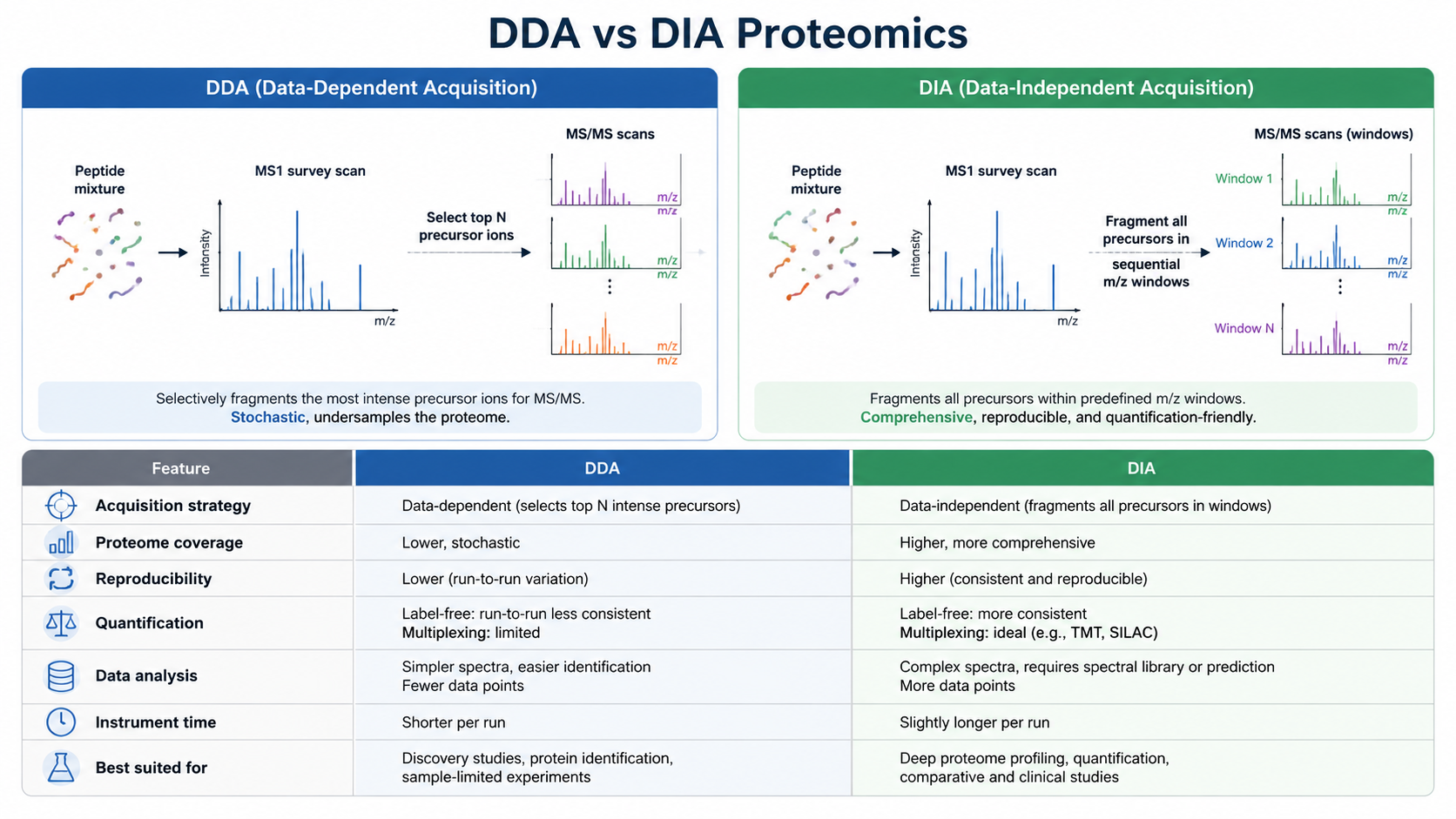
QC and Interpretation
Normalize; PCA for batch checks; differential testing; GO/KEGG/Reactome and PPI networks.
FAQ
What FDR is standard?
1% peptide/protein FDR is common in discovery studies.
Conclusion
Standardize each transition and bottom-up data support durable biological conclusions.
How to order?

