





Peptide Sequencing When Interpretation Fails: A Practical Troubleshooting Guide
- MS/MS spectra show weak or missing b-ion and y-ion series
- only a partial sequence tag is recovered from the spectrum
- database search returns low-scoring or conflicting peptide-spectrum matches
- co-eluting peptides produce chimeric or ambiguous spectra
- synthetic peptide verification fails due to unexpected mass or sequence differences
- modified residues shift fragment masses and complicate manual review
- the project deadline requires confirmed sequence before the next experimental step
- assigned peptide sequence with confidence annotation
- annotated MS/MS spectrum with major fragment assignments
- notes on ambiguous residues or incomplete ion series
- comparison against target sequence for verification projects
- recommendations for repeat acquisition or orthogonal confirmation
- Repeat LC-MS/MS with higher load or alternative fragmentation when spectral quality was borderline
- Intact mass or HPLC comparison when synthetic verification is the primary goal
- Orthogonal peptide mapping when the peptide must be placed in a larger protein context
- Repeat synthesis and purification when the sample itself appears compromised
Introduction
A peptide sequencing project can fail quietly. The LC-MS/MS run may complete without error, the precursor mass may look reasonable, yet the MS/MS spectrum shows weak fragmentation, the sequence tag is incomplete, or the database match score is too low to support a confident call. For teams preparing synthetic peptide release, unknown peptide identification, or biologics documentation, this outcome creates immediate delay.
MS/MS-based sequence determination usually fails for practical reasons rather than because the method itself is unsuitable. Low sample purity, insufficient peptide amount, poor ionization, co-eluting contaminants, incorrect digestion or handling, and overly complex mixtures can all reduce sequence confidence before interpretation begins. Repeating the same submission without reviewing sample quality or acquisition settings often produces the same ambiguous result.
When peptide sequence recovery stalls, the priority is to determine whether the problem lies in the sample, the separation, the MS/MS acquisition, or the original project assumptions. If your team is troubleshooting a failed peptide sequencing attempt or preparing a low-purity synthetic peptide for the first time, MtoZ Biolabs can Assess peptide sample readiness and recommend next steps before material is resubmitted.
Common Pain Points After Failed Peptide Identification
Researchers often seek help after encountering one or more of the following issues:
These problems are common in crude synthetic products, complex digest mixtures, natural product fractions, and samples stored improperly before analysis. The issue is often not whether LC-MS/MS sequence analysis is possible in principle. It is whether the current material and acquisition data can support reliable sequence assignment.
Why Peptide Sequencing May Fail
Before resubmitting samples or switching methods, review the most common failure points.
1. Low Purity or Co-Eluting Contaminants
Salts, truncations, deletion sequences, and unrelated peptides in the same fraction can suppress ion intensity or create mixed MS/MS evidence.
2. Insufficient Peptide Amount or Poor Ionization
Very low sample load may yield precursor signal without usable fragment ion series.
3. Incomplete or Noisy Fragmentation
Some sequences fragment poorly due to amino acid composition, internal labile bonds, or suboptimal collision energy.
4. Sample Handling and Storage Issues
Oxidation, deamidation, or hydrolysis can shift masses and create unexpected fragment patterns.
5. Over-Reliance on Database Search Alone
Weak matches may reflect an unknown sequence, a modified peptide, or a non-standard residue rather than instrument failure.
6. Incorrect Project Assumptions
A team may expect full L/I discrimination, complete modified-site localization, or protein-level answers from a single-peptide experiment.
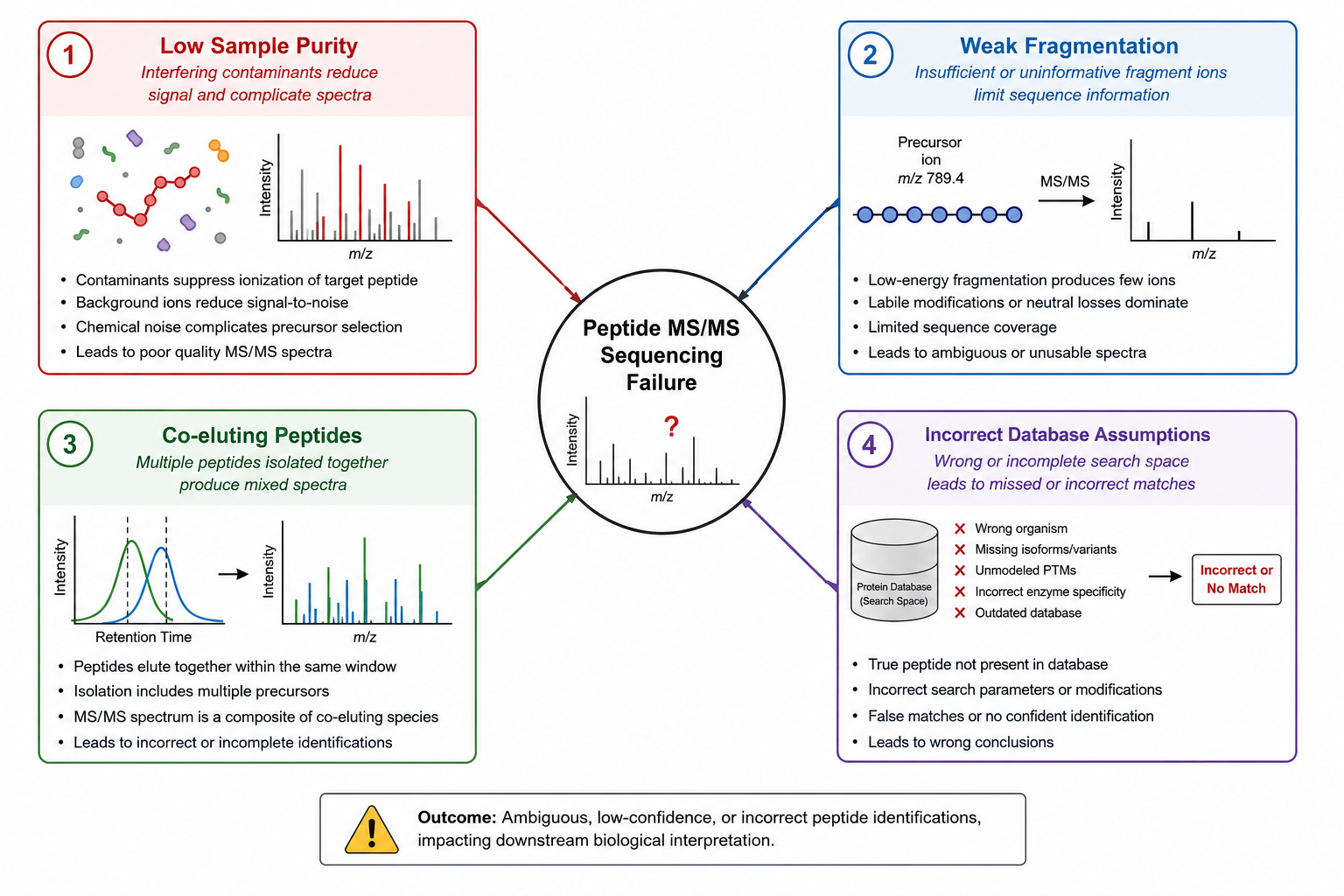
Figure 1. Failed peptide sequence recovery often reflects sample quality, spectral completeness, or interpretation complexity rather than method limitation alone.
Related Services
| Recommended Service Direction | |
|---|---|
| Need MS/MS-based peptide sequence determination | |
| Need de novo sequence for unknown peptides | |
| Need synthetic peptide sequence verification | |
| Need peptide identification without full sequencing | |
| Need peptide mapping against a known protein | |
| Need broader peptide MS identification support |
Step-by-Step Recovery Guide
When sequence recovery fails, use a structured review rather than repeating the same workflow.
Step 1: Confirm Sample Purity and Identity
Review HPLC traces, intact mass data, or prior QC results if available. If the sample contains multiple major components, additional purification may be required before MS/MS interpretation can succeed.
Step 2: Assess Peptide Amount and Preparation
Verify that enough material was submitted for the requested analysis. Repeat desalting, concentration, or cleanup if salts or buffer components may have suppressed ionization.
Step 3: Review MS/MS Acquisition Quality
Inspect precursor intensity, signal-to-noise, and the completeness of major fragment ion series. Weak spectra may require higher sample load, alternative fragmentation mode, or repeat LC-MS/MS acquisition.
Step 4: Re-evaluate Database Search Settings and Modifications
Step 5: Plan Validation or an Alternative Route
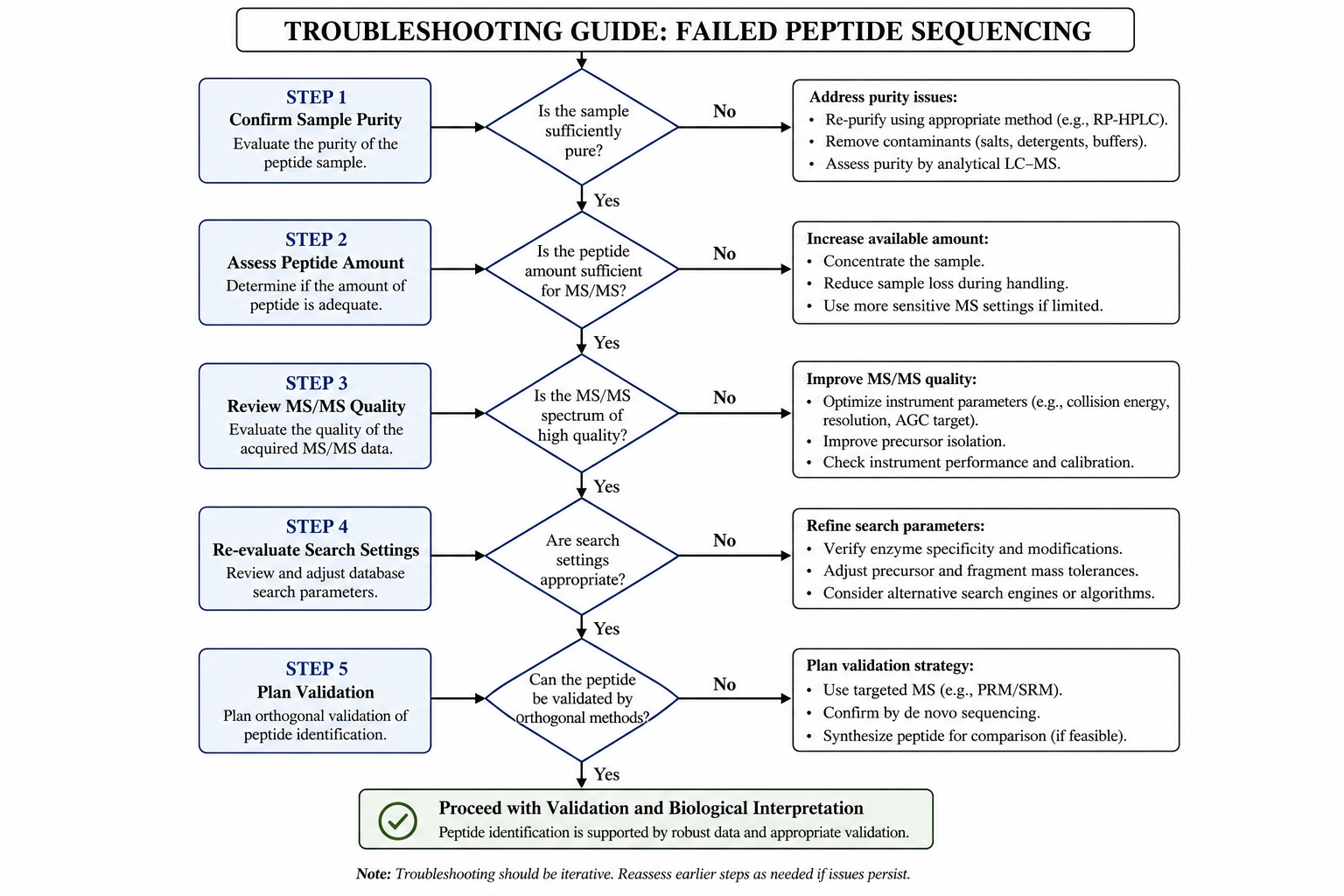
Figure 2. A structured troubleshooting path reduces unnecessary resubmissions and shortens sequence recovery time.
Peptide Sample Requirements
Sample quality is often the highest-leverage factor in successful MS/MS sequence assignment.

Figure 3. Feasibility review before shipment improves spectral quality and interpretation confidence.
For shipping, follow the provider's guidance on lyophilized versus liquid submission, cold-chain requirements, and metadata needs. Include target sequence, expected modifications, purity information, and any prior QC data when available.
Higher-purity material generally produces cleaner MS/MS spectra and faster manual review. If only crude material is available, communicate that clearly during feasibility review so the provider can recommend purification or scaled-up submission before LC-MS/MS analysis begins.
Expected Results and Validation Methods
A successful peptide recovery project should deliver more than a tentative sequence call.
Expected outputs may include:
Validation options depend on project goal:
Key Cautions
Do not assume that a precursor mass match alone confirms sequence identity. Isomeric or near-isobaric peptides can share precursor masses while differing in sequence.
Do not treat a low-scoring database hit as a confirmed sequence. Weak peptide-spectrum matches require manual spectrum review or de novo interpretation.
Do not skip metadata. Target sequence, modification expectations, and purity history help the analysis team choose the right search and review strategy.
Do not expect a single weak spectrum to support regulatory-grade verification without repeat evidence or orthogonal confirmation.
Frequently Asked Questions
1. Should I resubmit the same sample without changes?
Only after reviewing purity, amount, and prior MS/MS quality. Repeating the same workflow on poor material rarely improves the outcome.
2. Can a crude synthetic peptide still be sequenced?
Sometimes, if the target peptide ion can be isolated and fragmented cleanly. Additional purification often improves success rates.
3. What if only a partial sequence tag is recovered?
Partial tags may still narrow possibilities, but full verification usually requires extended ion series or repeat analysis.
4. Can I switch to PMF if MS/MS sequencing fails?
5. How can I reduce resubmission delays?
Submit the highest-purity material available, provide complete metadata, and request feasibility review before shipping.
Conclusion
Failed peptide sequencing is often a signal that sample purity, spectral quality, or interpretation strategy needs attention before sequence assignment can succeed. By reviewing preparation quality, MS/MS completeness, and project metadata before resubmitting material, teams can avoid repeated delays and obtain sequence evidence that supports the next experimental decision.
For projects where automated interpretation stalls, manual de novo review often resolves cases that search software alone cannot confidently assign. Providing target sequence context, expected modifications, and prior QC data helps the service team choose the fastest recovery route.
When peptide recovery fails or sample material is limited, MtoZ Biolabs can Plan a recovery workflow , . Contact the technical team to review sample status and the fastest path to usable peptide sequence data.
How to order?

