





Peptide Sequencing vs Database Search: Choosing the Right Peptide Identification Route
- the peptide sequence is unknown or uncertain
- synthetic verification requires independent spectrum evidence
- database search returns weak or conflicting matches
- modified or non-standard residues complicate automated identification
- documentation standards require annotated MS/MS support
- a reliable protein reference exists
- the goal is protein identification or coverage confirmation
- the sample is a standard digest with expected peptides
- speed and throughput matter more than manual spectrum review
- a reference sequence already exists
- the goal is coverage confirmation across a protein rather than discovery of one unknown peptide
- the peptide is high value
- regulatory or publication standards require orthogonal support
Introduction
Peptide identification projects rarely fail because teams lack mass spectrometry capability. They fail because the team chooses an identification route that does not match the sample type, reference availability, or required level of evidence. A synthetic peptide may need direct verification against a target design. A digest fraction may contain a peptide with no reliable database entry. A QC workflow may only require confirmation that observed masses match an expected protein sequence.
Researchers often compare MS/MS-based sequence determination, database-assisted identification, peptide mass fingerprinting, and Edman degradation when they need peptide-level answers. These methods can all produce useful data, but they begin from different assumptions and answer slightly different questions. Choosing the wrong route can waste time, consume limited material, and still leave the project without a sequence suitable for the next decision.
The central decision is not which platform is universally better. It is which method best matches the reference available, the sample complexity, and the downstream use of the recovered sequence. If your team is deciding between de novo spectrum interpretation and database search, MtoZ Biolabs can Compare peptide identification routes before samples are prepared or submitted.
Common Decision Scenarios
Method selection usually starts with one of four scenarios:
1. Unknown Peptide Sequence
The team has MS/MS data but no reliable reference for the peptide.
2. Synthetic Peptide Verification
A target sequence exists and must be confirmed in the purified product.
3. Protein Digest Identification
Peptides from a known or expected protein must be identified efficiently.
4. Documentation or Dispute Resolution
A prior database match is weak and requires independent spectrum review.
These scenarios lead to different method priorities. Unknown peptides usually favor de novo peptide sequencing or manual MS/MS interpretation. Known-protein digests often favor database search or PMF when coverage confirmation is the goal.
Related Services
| Customer Need | Recommended Service Direction |
|---|---|
| Need MS/MS-based peptide sequence determination | |
| Need de novo sequence for unknown peptides | |
| Need synthetic peptide sequence verification | |
| Need peptide identification without full sequencing | |
| Need peptide mapping against a known protein | |
| Need broader peptide MS identification support |
Key Comparison Dimensions
A useful comparison should focus on sample fit and project goal rather than generic platform preference.
| MS/MS Sequencing / De Novo Interpretation | Database Search / PMF | |
|---|---|---|
| Starting assumption | Sequence may be unknown | Reference protein or peptide set exists |
| Reference requirement | No | Yes, for efficient ID |
| Best for unknown peptides | Strong fit | Limited fit |
| Evidence type | Spectrum-derived sequence | Peptide-spectrum match or mass list match |
| Typical turnaround | Often longer for manual review | Often faster for standard digests |
| Ideal deliverable | Annotated peptide sequence from MS/MS |
Peptide Sequencing and De Novo MS/MS Interpretation
MS/MS-based sequence determination, in the strict sense, uses fragmentation data to determine residue order directly from observed b-ion and y-ion series. It is the preferred route when the peptide sequence is unknown, when synthetic verification requires independent spectrum evidence, or when database search returns ambiguous results.
Strengths include direct spectrum-level evidence, independence from public database completeness, and strong fit for unknown peptides, modified sequences, and non-standard samples. Manual or expert-assisted interpretation can resolve cases where automated search scores remain low.
Limitations include dependence on spectral quality, longer review time for difficult spectra, and possible L/I ambiguity without additional methods. Complex mixtures may require better separation before reliable interpretation is possible.
or
Database Search and Peptide Mass Fingerprinting
Database-assisted identification compares experimental MS/MS spectra or peptide mass lists against a reference sequence database. Peptide Mass Fingerprinting Analysis Service uses measured peptide masses from a digest to match a protein entry without full MS/MS sequencing of every peptide.
Strengths include speed, scalability, and strong performance for standard digests when the correct reference is present. For routine protein identification, database search is often the most efficient first step.
Limitations include dependence on database completeness and match quality. Unknown peptides, proprietary sequences, unexpected modifications, and weak scores may require de novo interpretation instead of another search iteration.
Edman Degradation and Orthogonal Confirmation
Edman degradation remains useful for N-terminal sequence analysis of purified peptides, especially when MS/MS data are incomplete or when a short N-terminal read provides sufficient confirmation. It is less commonly used as the primary route for long or modified peptides but can complement LC-MS/MS evidence in dispute cases.
Some projects combine methods strategically. Database search can identify most peptides in a digest, while targeted MS/MS sequencing resolves one critical unknown or modified peptide.
The best design depends on project goal and the evidence standard required for the next decision.
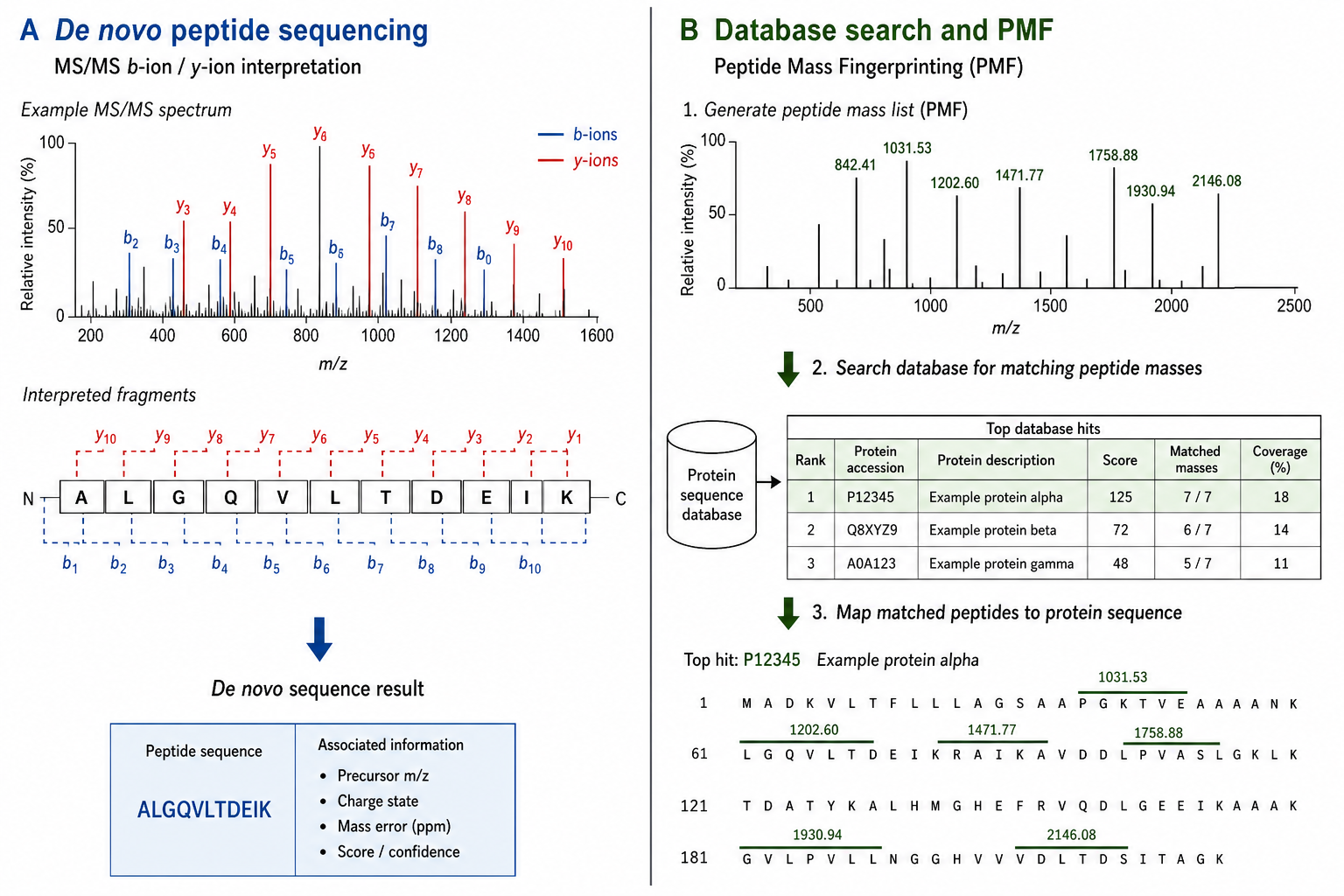
Figure 1. Reference availability and evidence requirements determine whether de novo MS/MS interpretation or database search is the better route.
Decision Recommendations by Project Goal
1. Choose MS/MS sequencing or de novo interpretation when:
2. Choose database search or PMF when:
3. Choose peptide mapping when:
4. Consider combined evidence when:
In-House vs Outsourced MS/MS Peptide Analysis
Some labs perform database search internally when peptide identification is routine and search infrastructure is established. However, difficult unknown peptides, synthetic verification disputes, and low-quality spectra still require expert MS/MS interpretation and manual review.
Outsourcing can reduce rework when sample purity is uncertain, modification patterns are complex, or the project requires report-ready deliverables on a fixed timeline. The tradeoff is vendor dependence, so teams should evaluate feasibility review, spectrum annotation quality, and communication before selecting a partner.
For occasional unknown-peptide projects, outsourced support is often more efficient than building occasional manual interpretation capability internally.
When comparing vendors, ask whether difficult spectra receive expert manual review rather than automated scoring alone. That distinction often determines whether ambiguous unknown peptides are resolved or returned as inconclusive search results.
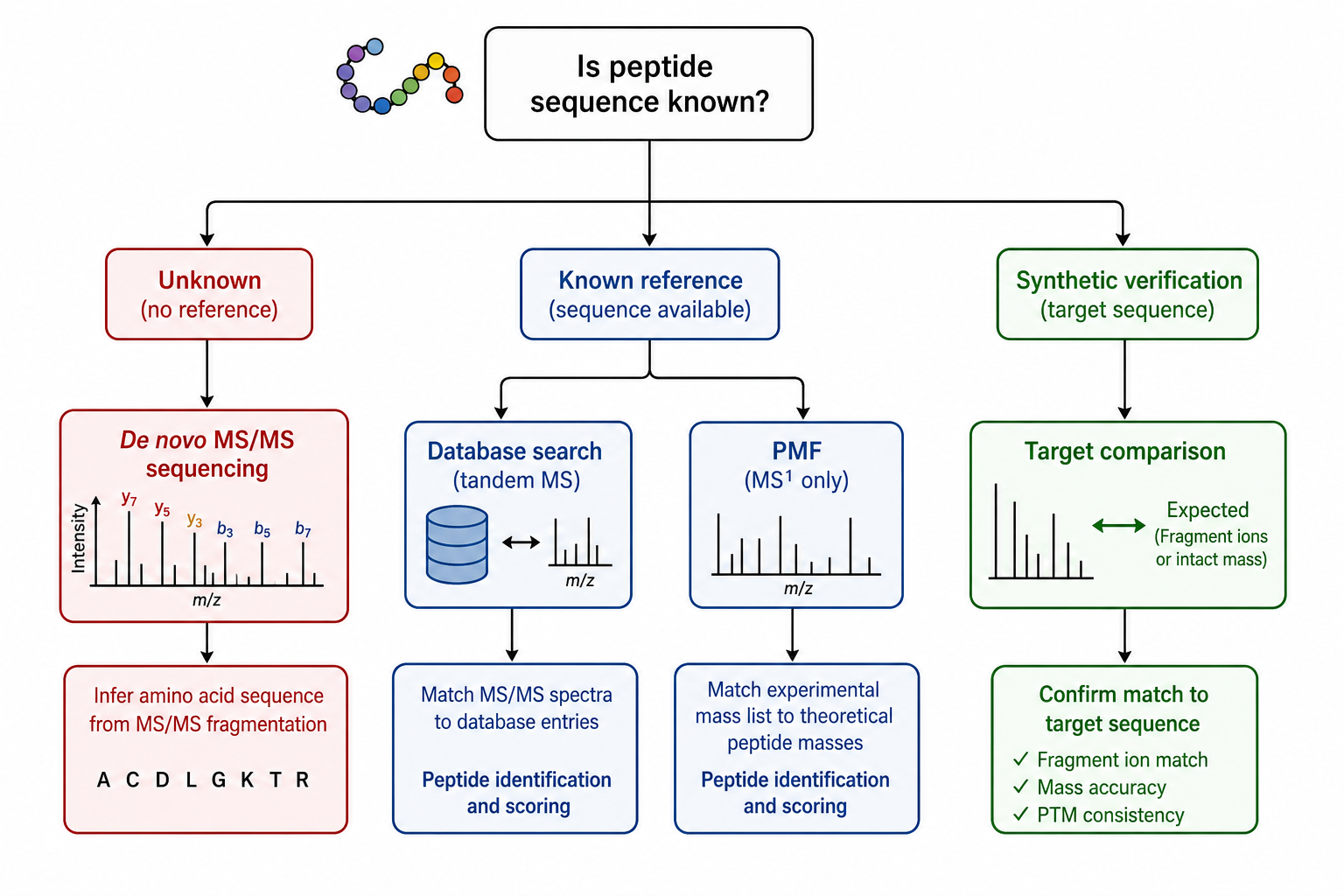
Figure 2. Unknown sequence status and reference availability determine the preferred peptide identification route.
Frequently Asked Questions
1. Is MS/MS sequencing always better than database search?
No. Database search is usually faster and more efficient when a reliable reference exists. De novo sequencing becomes preferable when the reference is missing, wrong, or unsupported by the data.
2. Can both routes be used in one project?
Yes. Many workflows use database search for routine peptides and targeted MS/MS sequencing for one unresolved peptide.
3. Does PMF replace full MS/MS sequencing?
Not for unknown peptides. PMF is most useful when a reference protein exists and measured masses are sufficient for identification.
4. What if my synthetic peptide fails database verification?
5. Can database search identify modified peptides?
Sometimes, if the modification is included in search parameters. Unexpected or complex modifications often require manual sequencing review.
Conclusion
Peptide sequencing and database search answer overlapping but distinct project needs. De novo MS/MS interpretation is usually the stronger choice when the peptide sequence is unknown or requires independent verification. Database search and PMF are the better first steps when a reliable reference exists and the goal is efficient protein identification or coverage confirmation. Method selection should begin with reference availability and evidence requirements, not platform preference alone.
Cell-based recovery is usually the stronger choice when viable cells or RNA remain available. Protein-level recovery is the better fallback when hybridoma stocks are gone but purified antibody remains. Method selection should begin with sample availability, not platform preference alone.
Match the identification route to sample type and project goal across , , and Contact the technical team to compare options before sample submission.
How to order?

