





Peptide Sequencing: Principles, Analytical Workflows, and Proteomics Applications
Introduction
Peptide sequence is often the first concrete structural answer a proteomics project needs. A synthetic peptide may require verification before downstream use. A fraction from enzymatic digestion may contain an unknown peptide that must be characterized before protein-level conclusions are made. A biopharmaceutical QC workflow may need to confirm that a critical fragment matches the expected primary structure.
The process is the determination of amino acid order from mass spectrometry data, most commonly by interpreting MS/MS fragmentation spectra produced during LC-MS/MS analysis. Unlike full protein sequencing, which assembles many overlapping peptides into a longer primary structure, peptide-level work focuses on one peptide ion at a time. Each spectrum reflects how a selected precursor breaks into fragment ions that encode residue order. Because interpretation depends on spectrum quality, sample purity, and whether a reliable reference exists, the analytical workflow must be chosen deliberately rather than by default.
Related Services
| Research Need | Recommended Service Direction |
| MS/MS-based peptide sequence determination | Peptide Sequencing Service |
| De novo sequence for unknown peptides | Peptide De Novo Sequencing Service |
| Synthetic peptide sequence verification | Verification Service of Synthetic Peptide Sequence |
| Peptide identification without full sequencing | Mass Spectrometry-Based Peptide Identification Service |
| Peptide mapping against a known protein | Peptide Mapping Service |
For projects where sample type, reference availability, or reporting depth is uncertain, MtoZ Biolabs can help evaluate whether database-assisted identification, de novo peptide sequencing, synthetic verification, or peptide mapping best fits the analytical goal.
What the Method Means in Practice
MS/MS-based sequence determination answers a focused question: what amino acid sequence best explains the fragmentation pattern observed for this peptide ion?
Database-assisted workflows assign spectra to known entries. The de novo route builds sequence evidence when the correct peptide sequence is unknown, proprietary, or potentially different from the expected design. Both routes rely on LC-MS/MS acquisition, but they differ in interpretation logic and reporting standards.
The method is especially relevant when researchers need to determine the sequence of an unknown peptide, verify a synthetic product, resolve ambiguous database matches, or document modified or truncated peptide forms. When a trustworthy protein reference already exists and the goal is coverage confirmation rather than discovery, peptide mapping may be sufficient. When the peptide sequence itself is unknown or disputed, MS/MS-based sequence determination becomes the primary route.
Core Principles of MS/MS-Based Sequence Determination
A typical workflow begins with sample introduction into liquid chromatography coupled to tandem mass spectrometry. Peptide ions are selected in the first mass analyzer, isolated, and fragmented. The resulting MS/MS spectrum contains product ions whose mass differences correspond to individual amino acid residues along the peptide backbone.
Analysts interpret characteristic b-ion and y-ion series to derive short sequence tags and extend them across the peptide. Software tools assist with peak assignment, but expert review remains important for sparse spectra, isobaric residues such as leucine and isoleucine, and modified amino acids that shift expected fragment masses.
Successful interpretation depends on several principles:
1. Precursor Quality
Sufficient precursor intensity and an informative charge state support cleaner fragmentation and more reliable ion series.
2. Fragment Ion Continuity
Consecutive b-ions or y-ions strengthen residue-order assignment. Gaps may indicate labile residues, internal fragmentation, or low-abundance peaks.
3. Redundant Evidence
Replicate spectra, complementary fragmentation modes, or overlapping sequence tags increase confidence.
4. Modification Awareness
Phosphorylation, oxidation, acetylation, and other modifications alter expected masses and must be considered during manual review.
Analytical Workflows in Proteomics
Two analytical paths dominate modern proteomics applications.
1. Database-Assisted Workflow
Experimental MS/MS spectra are matched against in silico fragment ions from a peptide or protein database. This approach is efficient for well-annotated proteomes, synthetic peptide confirmation against an expected sequence, and large-scale identification projects. Performance depends on database completeness, search parameters, and spectrum quality.
2. De Novo Workflow
Sequence tags are derived directly from fragment ions without requiring a prior database match. This path fits unknown peptides, proprietary sequences, engineered variants, and cases where database search returns weak or conflicting assignments. It usually requires more manual interpretation than routine search-based identification.
Many projects use a hybrid strategy: database search for the majority of spectra, followed by de novo analysis of unmatched or biologically important ions. This preserves throughput while still recovering sequence evidence that reference matching alone would miss.
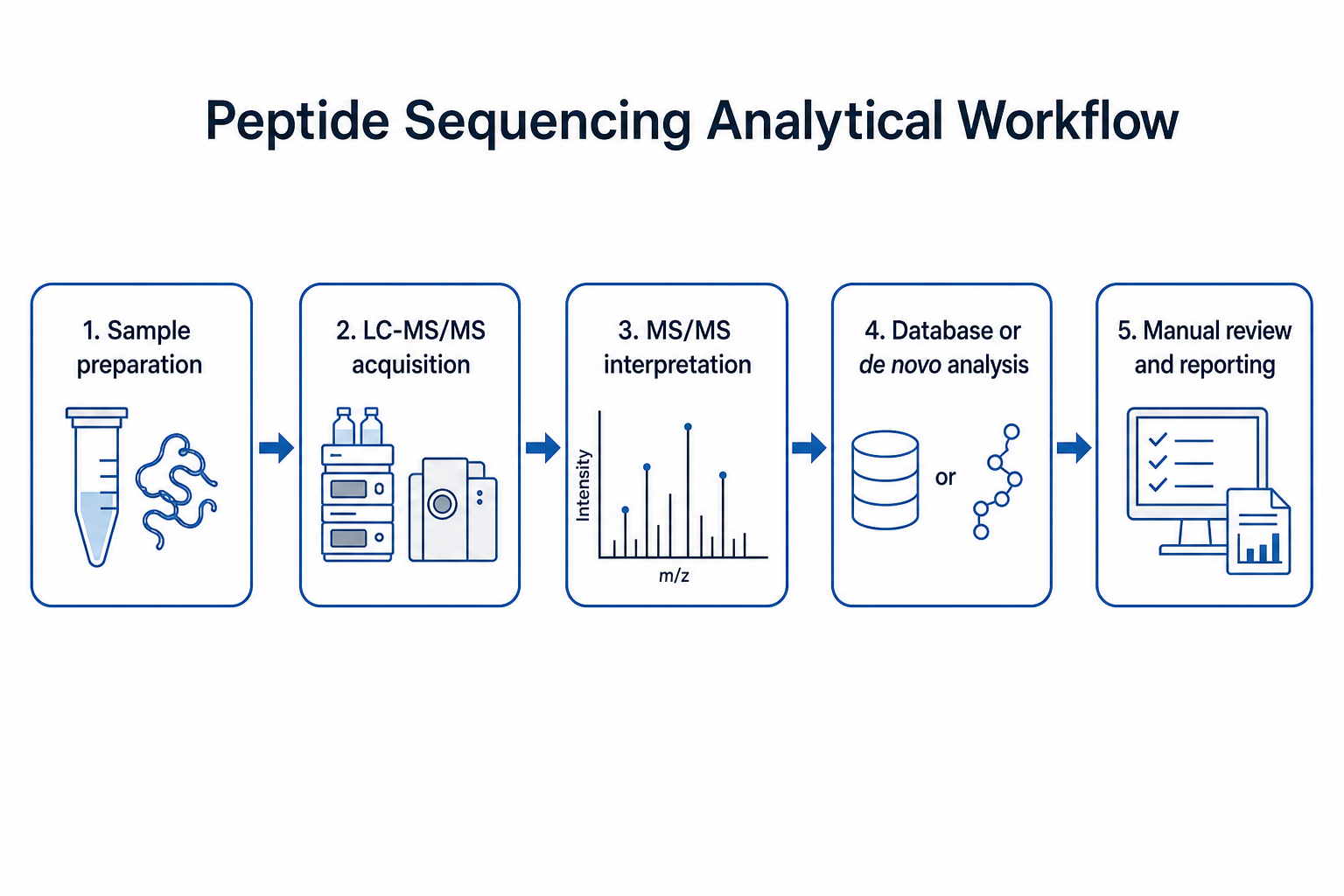
Figure 1. Analytical workflow from sample preparation through MS/MS interpretation and reporting
Workflow choice should be defined before acquisition when possible. A discovery proteomics study on a well-annotated sample may prioritize database search. A synthetic verification or unknown peptide project may require de novo interpretation from the start.
Sample Requirements
Sample quality strongly influences whether dependable peptide sequence calls are feasible. Cleaner input generally produces sharper spectra and more efficient interpretation.
| Sample Factor | Recommended Condition | Why It Matters |
| Sample format | Purified peptide, desalted digest fraction, or enriched LC fraction | Reduces spectral complexity and improves target ion selection |
| Purity | Single dominant peptide or clearly separated fraction | Lowers chimeric spectra and mixed precursor interference |
| Peptide amount | Enough for repeat LC-MS/MS when needed | Limited input reduces replicate support and method optimization |
| Sample matrix | Minimal salts, detergents, or polymers | Matrix components can suppress ionization and fragment quality |
| Modifications | Expected PTMs or chemical labels disclosed | Modifications affect fragmentation and manual interpretation |
| Background information | Expected sequence, organism, or synthesis record if available | Helps choose database-assisted or de novo analysis |
Short, highly pure peptides with strong fragmentation often yield fast, high-confidence results. Longer peptides, complex mixtures, or heavily modified sequences may require repeat runs, alternative fragmentation modes, or complementary analyses.
Technical Challenges and Current Limits
The method is powerful, but several technical challenges limit confidence if they are ignored.
1. Incomplete Fragmentation
Weak or sparse b-ion and y-ion series reduce confidence in sequence assembly Long peptides, poor precursor intensity, and suboptimal collision energy all contribute to this problem.
2. Isoleucine and Leucine Ambiguity
Many workflows cannot distinguish I and L by mass alone. Additional evidence or orthogonal methods may be needed for unambiguous assignment in critical regions.
3. Post-translational Modifications
Phosphorylation, glycosylation, oxidation, and other modifications can complicate peak assignment and residue ordering.
4. Chimeric or Mixed Spectra
Co-isolated precursors in complex samples can produce misleading fragment patterns that look plausible during automated scoring.
5. Low-abundance Peptides
Weak signals may require additional fractionation, targeted acquisition, or longer LC gradients to obtain interpretable MS/MS data.
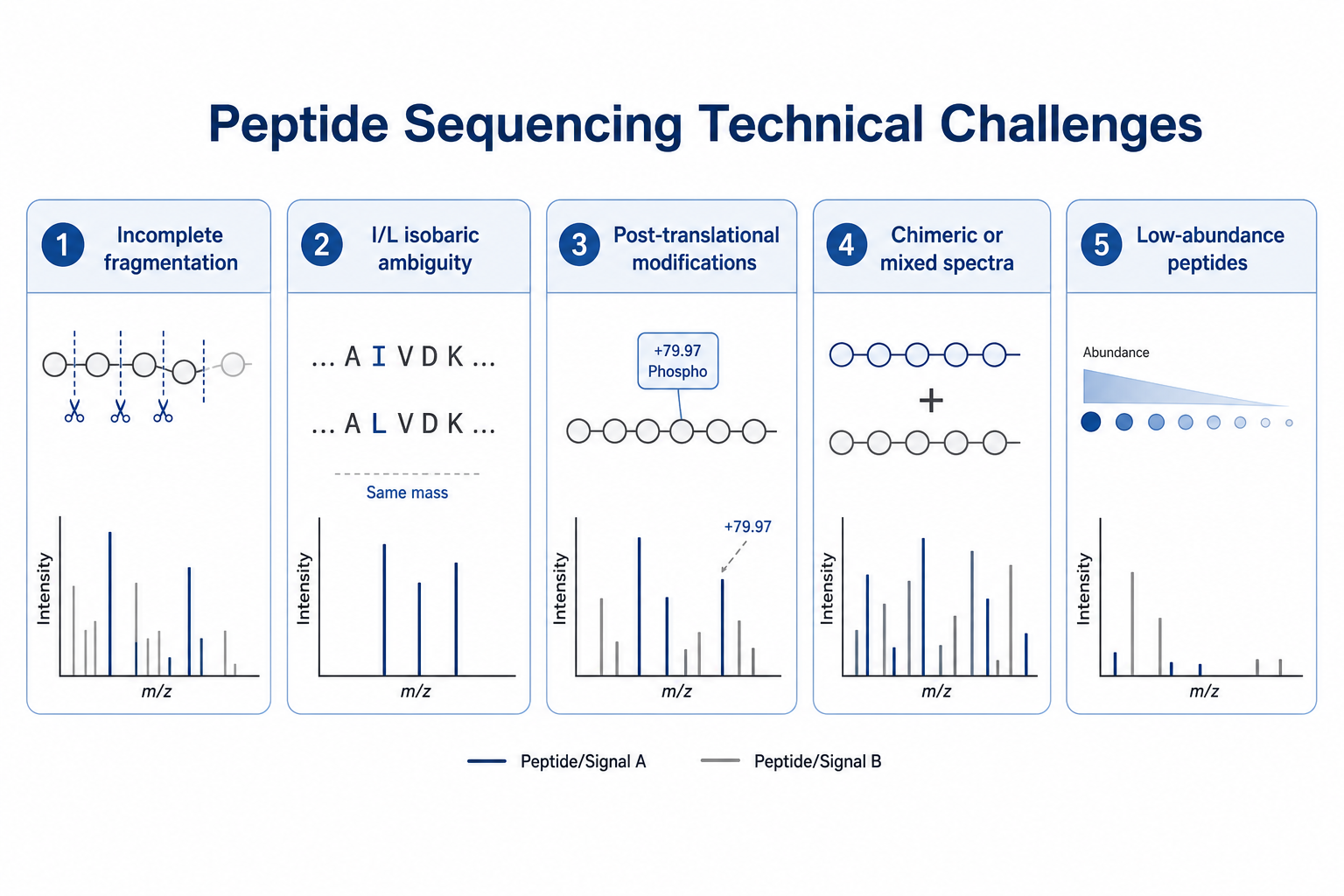
Figure 2. Common technical challenges that limit peptide sequence confidence
These limits explain why reporting should distinguish high-confidence sequence calls from tentative assignments. A peptide sequence without spectrum support, ambiguity flags, or modification notes can create a false sense of certainty in downstream decisions.
Expected Outputs and Reporting Depth
Reporting depth should match the biological or commercial decision behind the project.
| Output Type | Typical Content | Best Used For |
| Confirmed peptide sequence | Assigned amino acid order with spectrum support | Synthetic verification and unknown peptide ID |
| Annotated MS/MS spectra | Key ions linked to residue assignments | Manual audit, publication, or QC review |
| Sequence tags | Short confirmed stretches from fragment series | Exploratory mapping and follow-up design |
| Ambiguity flags | I/L positions, low-confidence residues, or gaps | Transparent reporting and validation planning |
| Modification annotation | Located PTMs or chemical adducts | Biopharmaceutical and modified peptide work |
| Method summary | Database, de novo, or hybrid interpretation path | Project documentation and comparability review |
A strong sequence report separates confident calls from tentative ones and indicates where repeat acquisition or orthogonal validation would most improve the evidence.
Proteomics Applications
The method supports several recurring proteomics use cases.
1. Unknown Peptide Identification
A peptide from digestion or fractionation can be sequenced to support protein inference, clone design, or homology analysis when database search is insufficient.
2. Synthetic Peptide Verification
Manufactured peptides can be checked against the intended design before biological use or release documentation.
3. Discovery Proteomics Support
Unmatched or low-scoring spectra from complex proteomes can be interpreted to recover sequence tags that expand identification beyond the reference database.
4. Biopharmaceutical QC
Critical peptide fragments from biologics can be verified for sequence accuracy, truncations, or unexpected processing.
5. Modified Peptide Characterization
Phosphopeptides, oxidized peptides, and other modified forms can be analyzed when modification-aware interpretation is required.
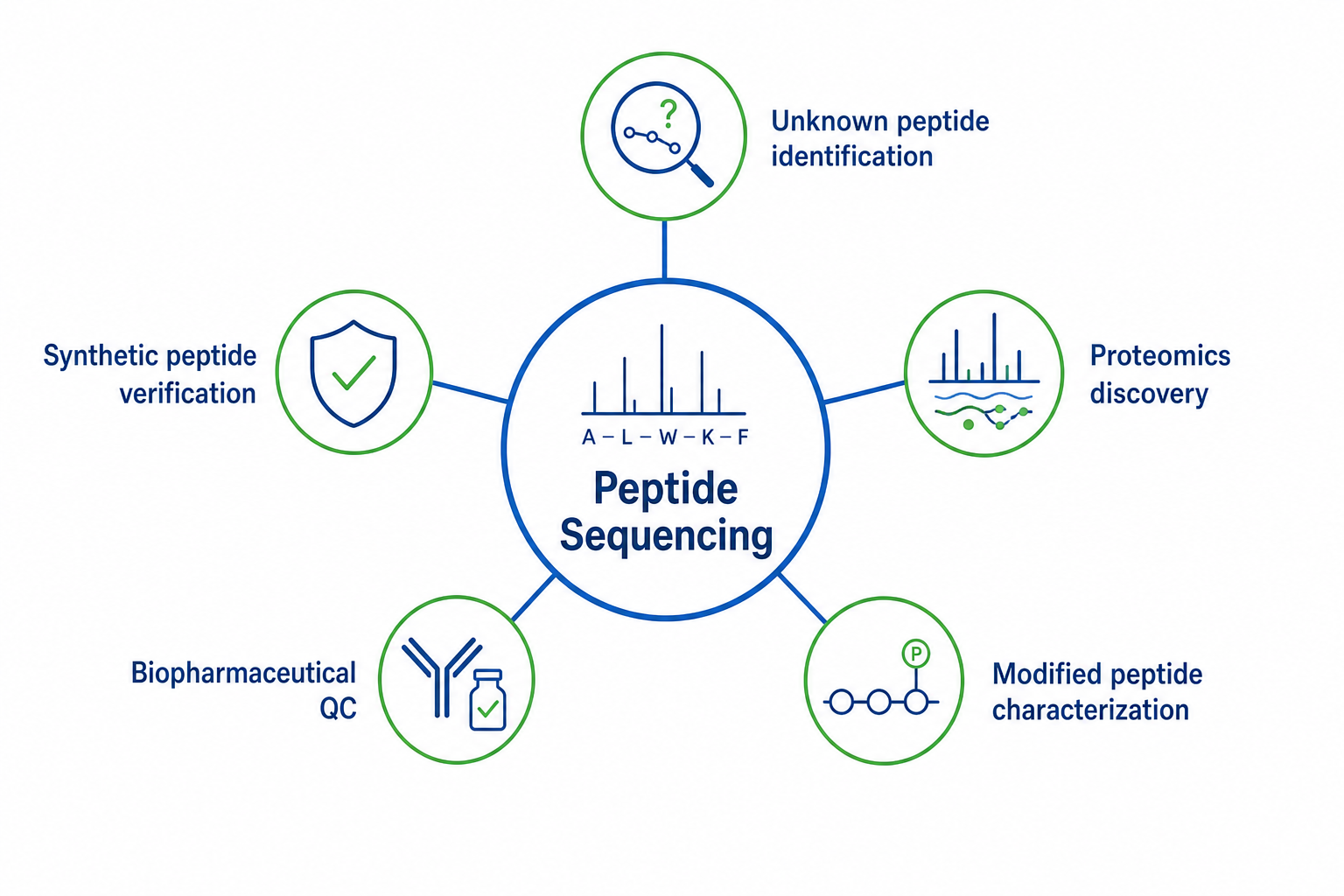
Figure 3. Proteomics applications supported by the method
In each application, the required confidence level should be defined early. A single strong spectrum may be enough for one exploratory question, while QC or regulatory documentation may require replicate support and annotated reporting.
Advantages and Current Limits
The main advantage is direct sequence evidence at the peptide level when identification alone is not enough. LC-MS/MS-based analysis can examine synthetic products, digested proteins, and fractionated mixtures with residue-level detail that intact mass measurement alone cannot provide.
Current limits are equally important. Not every spectrum supports a full sequence call. Manual interpretation adds time and expertise. Highly complex mixtures, chimeric precursors, and isobaric residues can reduce confidence. The workflow is therefore most effective when combined with realistic project scoping, clean sample preparation, and transparent reporting standards.
Future Outlook
MS/MS sequencing workflows are likely to continue integrating database search, de novo interpretation, electron-based fragmentation, chemical derivatization, and targeted MS. Improvements in mass accuracy, acquisition speed, and interpretation software will help, but dependable sequence reporting will still depend on spectrum quality and expert review for high- stakes applications.
The most useful future model is not full automation of every peptide call. It is a layered workflow in which database-assisted identification handles routine coverage while de novo peptide sequencing and manual review address the spectra that carry the greatest scientific or commercial weight.
Frequently Asked Questions
1. What is the difference between peptide identification and sequence determination?
Peptide identification assigns a spectrum to a known database entry. Sequence determination derives or verifies amino acid order from the MS/MS spectrum itself, especially when the sequence is unknown or must be independently confirmed.
2. When is de novo peptide sequencing needed?
De novo interpretation is needed when the correct sequence is absent from the database, when the peptide may differ from the expected design, or when database search produces ambiguous or weak matches.
3. What sample types are most suitable?
Purified peptides, synthetic products, and well-separated LC fractions are commonly suitable. Complex mixtures are more difficult unless the target precursor can be cleanly selected.
4. Can LC-MS/MS determine modified peptide sequences?
Yes, when modification-aware interpretation is used. Modifications shift fragment masses and should be considered during acquisition design and manual review.
5. How does the method support proteomics discovery?
It helps recover sequence tags from unmatched spectra, verify critical peptides in complex datasets, and extend identification beyond standard database coverage in poorly annotated or variant-containing samples.
Conclusion
The workflow provides a practical route to amino acid sequence evidence at the peptide level through LC-MS/MS acquisition and MS/MS interpretation. It depends on sample quality, choice of database-assisted or de novo analysis, manual review, and transparent reporting of ambiguous residues and modifications. It is widely used for unknown peptide identification, synthetic verification, discovery proteomics support, biopharmaceutical QC, and modified peptide characterization.
For projects that need dependable peptide sequence reporting beyond routine identification, contact MtoZ Biolabs to discuss peptide sequencing, de novo analysis, synthetic verification, or an integrated LC-MS/MS analytical workflow.
How to order?

