





Peptide Sequencing Methods Compared: De Novo Analysis vs Database Search
Introduction
Most proteomics workflows use reference-based search as the default route for peptide assignment. The method is efficient, scalable, and well supported when a high-quality reference proteome or expected sequence is available. However, many peptide-level projects do not fit that assumption. A synthetic product may need independent verification. A digest fraction may contain a peptide absent from the database. A modified or variant-containing peptide may score poorly against the expected entry. In these cases, researchers must decide whether reference matching is still sufficient or whether de novo analysis is required to recover the true peptide sequence.
The choice is not about which peptide sequencing methods are more advanced. Reference matching asks which known sequence best explains an MS/MS spectrum. De novo analysis asks what sequence the spectrum itself supports. Selecting the wrong path can waste instrument time, produce false confidence, or miss the biologically important peptide entirely.
Related Services
| Research Need | Recommended Service Direction |
| MS/MS-based peptide sequence determination | Peptide Sequencing Service |
| De novo sequence for unknown peptides | Peptide De Novo Sequencing Service |
| Synthetic peptide sequence verification | Verification Service of Synthetic Peptide Sequence |
| Peptide identification in annotated proteomes | Mass Spectrometry-Based Peptide Identification Service |
| Peptide mapping against a known protein | Peptide Mapping Service |
When the best peptide-level approach is unclear, MtoZ Biolabs can help evaluate whether reference-based analysis, de novo peptide sequencing, synthetic verification, or a hybrid workflow best matches the sample, reference availability, and reporting goal.
When Researchers Face This Decision
The comparison usually appears at a specific decision point. A reference-based search may return no confident match for an important LC fraction. A synthetic peptide may need verification beyond a simple precursor mass check. A biopharmaceutical QC sample may show a peptide that does not fully align with the expected sequence. A metaproteomics project may contain peptides from poorly annotated organisms.
In each case, the practical question is the same: does the study need to match a known reference, or does it need to derive sequence that the database does not contain? That question should be answered before committing MS time, sample preparation effort, and data analysis strategy.
Four Comparison Dimensions That Matter Most
A useful comparison should focus on decision-relevant differences rather than generic descriptions. Four dimensions matter most for peptide-level projects: reference availability, study goal, throughput needs, and required sequence certainty.
1. Reference Availability
Reference-based search depends on a suitable peptide or protein sequence database, or on a supplied expected sequence. If the true peptide differs because of synthesis error, mutation, processing, or poor annotation, matching becomes weak or misleading. De novo analysis is designed for cases where the correct sequence cannot be assumed in advance.
2. Study Goal
Discovery proteomics on well-annotated samples favors reference matching. Unknown peptide identification, synthetic verification, variant confirmation, and independent sequence proof often require de novo interpretation or a hybrid strategy.
3. Throughput and Scale
Reference-based search is better suited to large-scale identification across many spectra and complex proteomes. De novo analysis is more labor-intensive and is usually targeted to unmatched spectra, purified peptides, or sequence-critical ions.
4. Sequence Certainty
Database matching can confidently assign peptides that fit known entries. De novo analysis can reveal novel or divergent sequence but often requires manual spectrum review, replicate support, and clear ambiguity labeling for dependable reporting.
Method Comparison at a Glance
The table below summarizes how the two main peptide sequencing methods differ on the dimensions most teams use during project planning.
| Method | Typical Use | Reference Needed? | Main Strength | Main Limitation |
| Reference- based search | Identification and expected-sequence confirmation | Yes, reliable reference or supplied sequence | Fast, scalable peptide assignment | Weak when sequence differs from database |
| De novo analysis | Unknown, synthetic, or disputed peptide sequence | No prior match required | Recovers sequence from thespectrum itself | Needs strong spectra and expert review |
| Hybrid workflow | Complex samples with both routine and critical peptides | Partial | Balances throughput and sequence recovery | Requires clear rules for follow-up analysis |
Hybrid approaches are common in practice. A project may use reference matching for most spectra, then apply de novo interpretation to unmatched, low-scoring, or biologically important ions.

Figure 1. Core differences between database search and de novo analysis at the peptide level
Researchers should compare peptide sequencing methods by the decision behind the project, not by software brand or lab habit alone. A workflow optimized for proteome coverage may be the wrong standard when the deliverable is independent verification of one synthetic peptide.
How Database Search Performs in Practice
Database search matches experimental MS/MS spectra to in silico fragment ions generated from a peptide or protein sequence database. Search engines score peptide-spectrum matches using mass accuracy, enzyme specificity, fragment coverage, and false discovery rate control.
The approach is often the right first choice for proteomics studies because it is fast, scalable, and statistically mature. It supports peptide assignment across complex samples, quantitative workflows, and modification searches when the underlying sequence is known. For model organisms, human samples, standard cell-line proteomes, and synthetic peptides with a supplied expected sequence, reference-based search remains the most efficient route.
The main weakness appears when the reference is wrong or incomplete. A missing variant, undocumented modification, synthesis mismatch, or absent database entry can prevent a valid spectrum from receiving a confident match. In those cases, the issue is often not poor MS data. The issue is that the database does not contain the peptide being measured.
How De Novo Analysis Performs in Practice
De novo analysis derives peptide sequence from the MS/MS spectrum itself. Fragment ions are interpreted as residue-level mass differences, and candidate sequences are assembled without requiring a prior database match. This makes the method valuable for unknown digest fractions, synthetic peptide verification, modified peptides, environmental samples, and any project where the measured sequence may differ from available references.
The de novo route is strongest when spectra are high quality and the target peptide is enriched or cleanly selected. It is weaker when spectra are sparse, mixtures are complex, or the project expects rapid high-throughput identification across an entire run without manual review. The method is therefore often used selectively rather than as a replacement for all database-search steps.
In many successful projects, de novo interpretation is applied after reference search fails or after unmatched high-quality spectra are prioritized. This focused use preserves efficiency while still recovering sequence evidence that would otherwise be lost.
Which Approach Fits Different Study Goals
The best choice depends on what the study must prove.
Choose reference-based search when the sample comes from a well-annotated source, the goal is broad peptide assignment or quantification, and the expected sequences are represented in the reference or supplied design file. Typical examples include cell-line proteomics, pathway studies, and synthetic peptide confirmation against a known target sequence.
Choose de novo analysis when the peptide sequence is unknown, proprietary, engineered, modified in an unexpected way, or likely to differ from available references. Typical examples include unknown LC fractions, disputed synthetic products, variant peptides, and sequence- critical QC ions.
Choose a hybrid workflow when most peptides can be identified by reference matching but a subset of high-value spectra require direct sequence derivation. This is common in biopharmaceutical QC, metaproteomics, and projects that combine discovery coverage with targeted sequence proof.
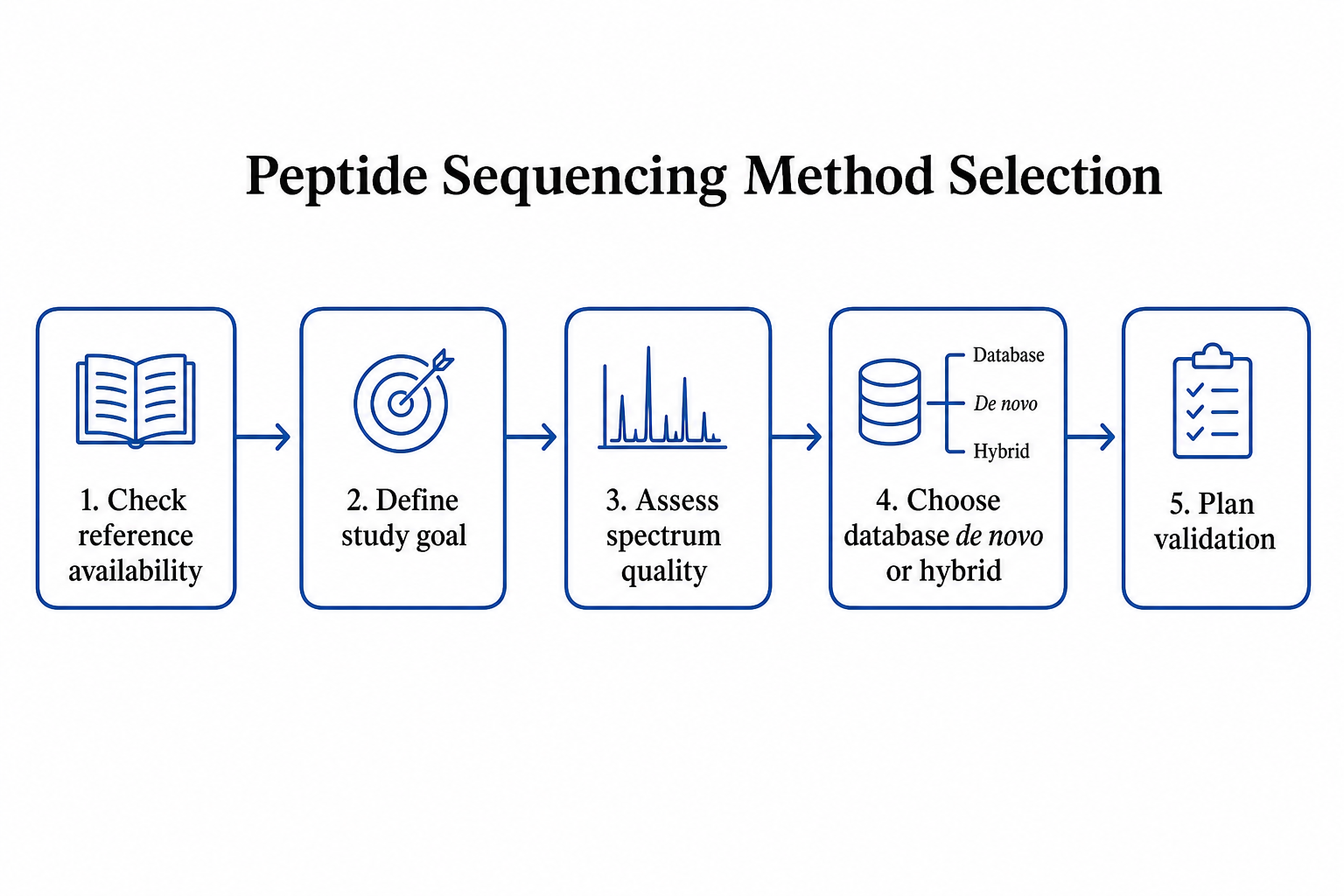 Figure 2. Decision flow for choosing reference matching, de novo analysis, or a hybrid workflow
Figure 2. Decision flow for choosing reference matching, de novo analysis, or a hybrid workflow
Reporting depth should be defined before method selection. If the project only needs protein names or peptide IDs, reference-based search may be enough. If the project needs residue-level confirmation, independent synthetic proof, or documentation for QC and publication, de novo analysis or confirmatory review should be planned from the start.
Research Goal and Method Fit
| Research Goal | Usually Best Starting Point | When to Add a Second Method |
| Synthetic peptide verification | Reference-based search against expected sequence | Add de novo analysis if mismatch ions or weak scores appear |
| Unknown peptide from a digest fraction | De novo analysis | Add reference search secondarily for homology context |
| Broad proteome discovery | Reference-based search | Re-analyze unmatched spectra with de novo tools |
| Modified peptide confirmation | Reference search with modification parameters | Add de novo review for ambiguous modified regions |
| Biopharmaceutical peptide QC | Hybrid workflow | Target low-scoring or unexpected peptides for manual review |
| Poorly annotated organism sample | Hybrid workflow | Prioritize unmatched spectra for de novo interpretation |
A strict either-or choice is not always necessary. The most efficient project design often uses reference matching for routine coverage and reserves de novo analysis for the spectra that carry the greatest scientific or commercial weight.
Limitations to Keep in Mind
Neither approach among common peptide sequencing methods is universally superior. Reference-based search is only as good as the reference database, search parameters, and modification settings. Incomplete references, poor enzyme specificity assumptions, and underestimated modification complexity can all reduce assignment quality. De novo analysis depends on spectrum quality and expert review. Ambiguous residues, chimeric spectra, and isoleucine or leucine uncertainty can limit confidence.
Researchers should also avoid comparing the two methods only by peptide count. Identification coverage and sequence proof are not the same deliverable. A workflow optimized for discovery throughput may be the wrong conclusion when the real need is independent verification of one critical peptide.
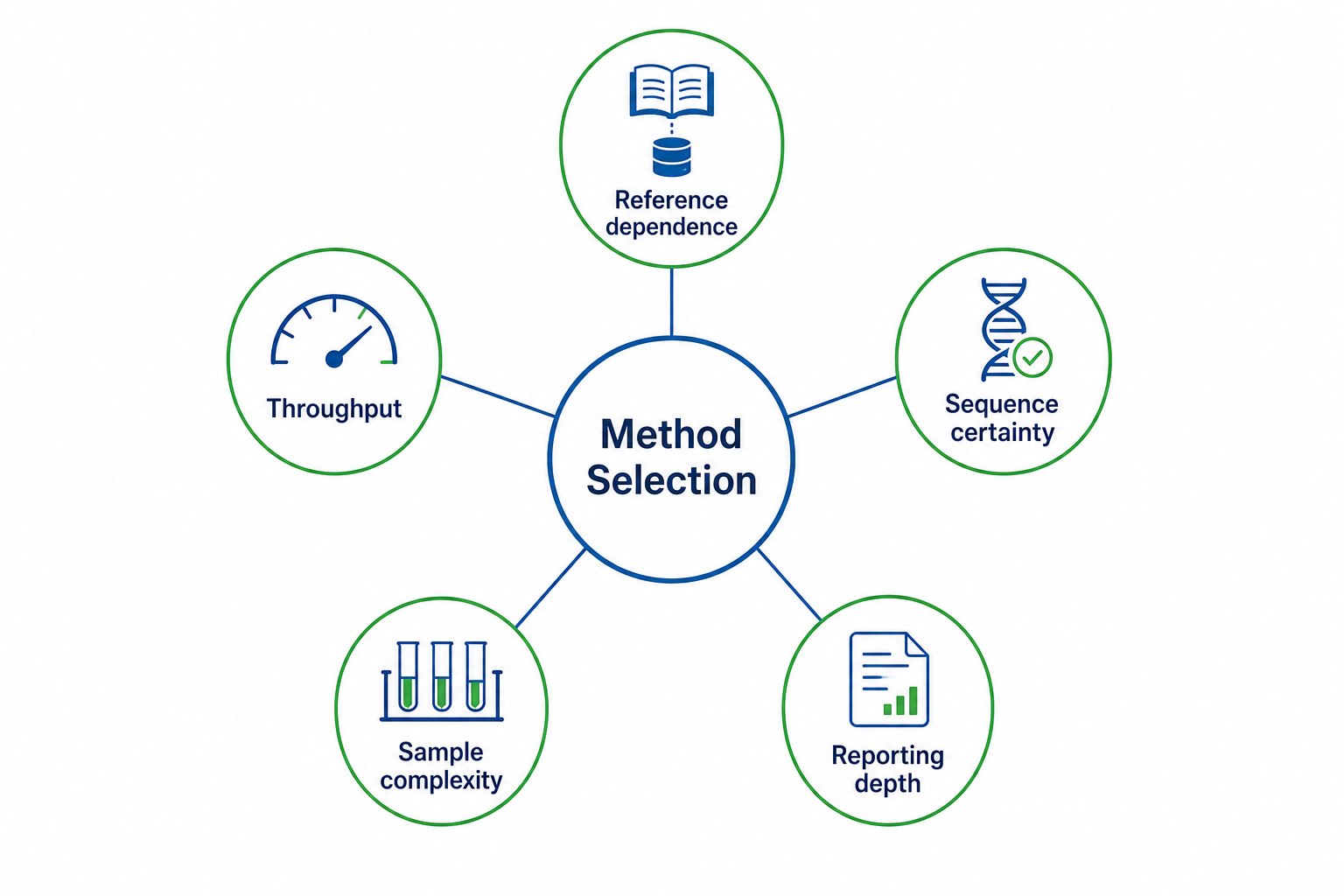
Figure 3. Key tradeoffs to weigh when selecting a peptide sequencing method
Before starting data analysis, confirm whether a reliable reference exists, whether the true peptide may differ from that reference, whether the goal is coverage or sequence proof, and whether the final report must support QC, publication, or regulatory use.
Frequently Asked Questions
1. Is de novo analysis better than database search for peptides?
Neither method is better in all cases. Database search is better for high-throughput identification when references are reliable. De novo analysis is better when the peptide sequence may be absent, incorrect, or intentionally different from the reference.
2. Can database search identify unknown peptides?
Database search can assign homologous peptides if related sequences exist in the database. If the peptide is truly novel or too divergent, de novo analysis is usually needed to derive the sequence directly from the spectrum.
3. When should a peptide project use both methods?
A hybrid workflow is useful when most spectra can be assigned by reference matching but a subset of unmatched, low-scoring, or biologically important ions require de novo interpretation.
4. Which method is faster?
Reference-based search is generally faster and more scalable for large datasets. De novo analysis requires more manual interpretation and is usually applied to selected spectra or purified peptide samples.
5. Which approach is better for synthetic peptide verification?
Synthetic verification often starts with reference matching against the expected sequence. If scores are weak or unexplained fragment ions remain, de novo analysis or manual spectrum review is often needed for dependable confirmation.
Conclusion
Peptide sequencing methods serve different analytical needs. Reference-based search fits discovery studies and expected-sequence confirmation when references are reliable. De novo analysis fits projects that require direct sequence derivation for unknown, synthetic, modified, or variant-containing peptides. Hybrid workflows often provide the best balance when only a subset of spectra needs sequence-level interpretation.
If your project sits at the boundary between identification and sequence proof, contact MtoZ Biolabs to discuss whether reference-based analysis, de novo peptide sequencing, synthetic verification, or a combined LC-MS/MS workflow is the right fit.
How to order?

