





Peptide Sequencing: How LC-MS/MS Determines Amino Acid Sequences from MS/MS Spectra
- determine the sequence of an unknown peptide from a digest or fraction
- verify that a synthetic peptide matches the intended design
- confirm sequence identity for patent, publication, or batch release documentation
- resolve ambiguous database matches with spectrum-level evidence
- characterize truncated, modified, or unexpectedly processed peptide products
- assigned peptide sequence with confidence annotation
- annotated MS/MS spectrum with major b-ion and y-ion assignments
- precursor m/z, charge state, and observed mass values
- notes on ambiguous residues, modifications, or incomplete ion series
- comparison against target sequence when verification is the project goal
- recommendations for repeat analysis or orthogonal confirmation if needed
Introduction
Peptide sequence information is often the first concrete answer a project needs. A synthetic peptide may require verification before animal studies. A purified fraction from digestion may contain an unknown peptide that must be identified before the parent protein can be characterized. A biopharmaceutical QC team may need to confirm that a critical peptide fragment matches the expected primary structure.
Peptide sequencing is the process of determining amino acid sequence from mass spectrometry data, most commonly by interpreting MS/MS fragmentation spectra produced during LC-MS/MS analysis. Unlike intact protein sequencing, which assembles many overlapping peptides into a full primary structure, peptide-level sequencing focuses on one peptide at a time. Each spectrum reflects how a selected precursor ion breaks into fragment ions that encode residue order.
Researchers use MS/MS-based sequence determination in proteomics, synthetic peptide QC, natural product analysis, and biologics characterization. The method is especially valuable when the peptide sequence is unknown, when a synthetic product must be verified against a target design, or when database search alone produces ambiguous matches. For teams evaluating whether a peptide sample can support reliable sequence determination, MtoZ Biolabs can Review LC-MS/MS project fit before samples are prepared or submitted.
What Question Does Peptide Sequencing Answer?
At its core, this workflow answers a focused question: what amino acid sequence best explains the MS/MS fragmentation pattern observed for this peptide ion?
Database-assisted peptide identification asks whether a known entry matches the experimental spectrum. De novo peptide sequencing asks what sequence the spectrum itself supports when no reliable reference is available or when independent verification is required.
The method is especially relevant when researchers need to:
When a high-confidence protein reference already exists and the goal is coverage confirmation rather than discovery, Peptide Mapping Service may be sufficient. When the peptide sequence itself is the unknown, LC-MS/MS-based sequence determination becomes the primary route.
Related Services
| Recommended Service Direction | |
|---|---|
| Need MS/MS-based peptide sequence determination | |
| Need de novo sequence for unknown peptides | |
| Need synthetic peptide sequence verification | |
| Need peptide identification without full sequencing | |
| Need peptide mapping against a known protein | |
| Need broader peptide MS identification support |
How Peptide Sequencing Works
The workflow typically begins with a purified peptide sample or a peptide-containing fraction from enzymatic digestion. The sample is introduced into a liquid chromatography system coupled to a tandem mass spectrometer. Peptide ions are selected in the first mass analyzer, isolated, and fragmented in the collision cell. The resulting MS/MS spectrum contains a series of product ions whose mass differences correspond to individual amino acid residues.
Analysts interpret characteristic b-ion and y-ion series to derive short sequence tags and extend them across the peptide backbone. Software tools can assist with peak assignment, but expert review remains important for low-abundance ions, isobaric residues such as leucine and isoleucine, and modified amino acids that shift expected mass patterns.
A typical LC-MS/MS workflow includes:
1. Feasibility Review of Sample Purity, Amount, and Project Goal
2. Sample Preparation and Optional Desalting or Fractionation
3. LC Separation and MS/MS Data Acquisition
4. Spectrum Interpretation and Sequence Tag Assembly
5. Manual Review of Ambiguous Regions and Modification Sites
6. Report Delivery with Annotated Spectra and Confidence Notes
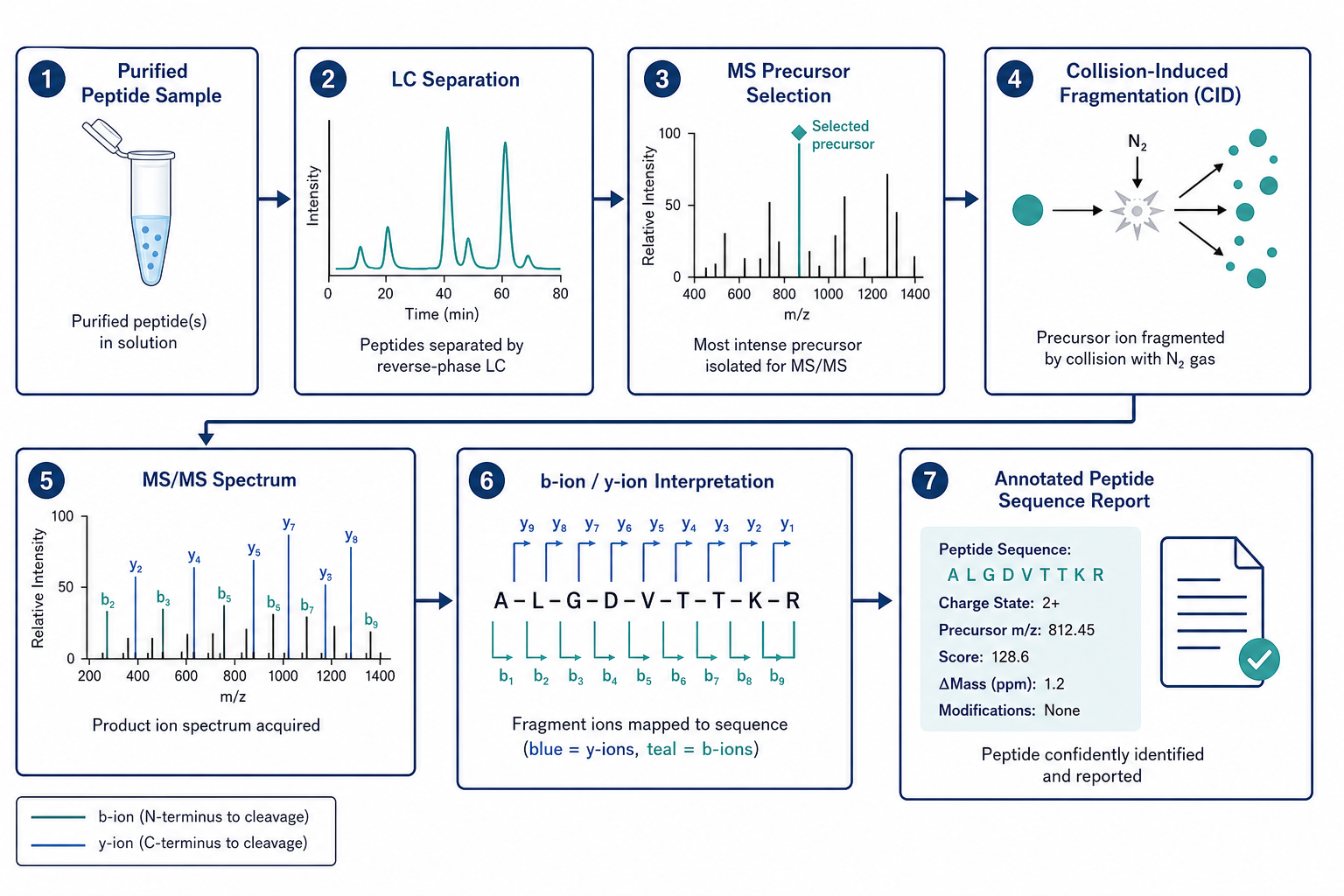
Figure 1. MS/MS-based sequencing converts fragmentation data into amino acid sequence evidence through LC-MS/MS acquisition and expert interpretation.
The quality of the final sequence depends on spectral quality, peptide length, sample purity, instrument resolution, and the presence of modifications. Short, highly pure peptides with strong fragmentation often yield fast, high-confidence results. Longer peptides, complex mixtures, or heavily modified sequences may require repeat runs, alternative fragmentation modes, or complementary analyses.
MS/MS Spectrum Interpretation Basics
Successful sequence determination depends on reading fragmentation patterns, not just identifying a precursor mass.
1. Precursor Ion Selection
The instrument isolates a peptide ion of interest based on m/z. Precursor charge state affects fragmentation behavior and interpretation strategy.
2. b-Ion and y-Ion Series
N-terminal fragment ions (b-series) and C-terminal fragment ions (y-series) provide complementary evidence for residue order. Gaps in a series may indicate labile residues, internal fragmentation, or low-abundance peaks.
3. Sequence Tags
Short confirmed sequence stretches anchor full peptide assembly. Longer tags and redundant ion series increase confidence.
4. Isobaric Residues and Modifications
Leucine and isoleucine cannot be distinguished by mass alone in many workflows. Phosphorylation, oxidation, acetylation, and other modifications shift expected fragment masses and require explicit review.
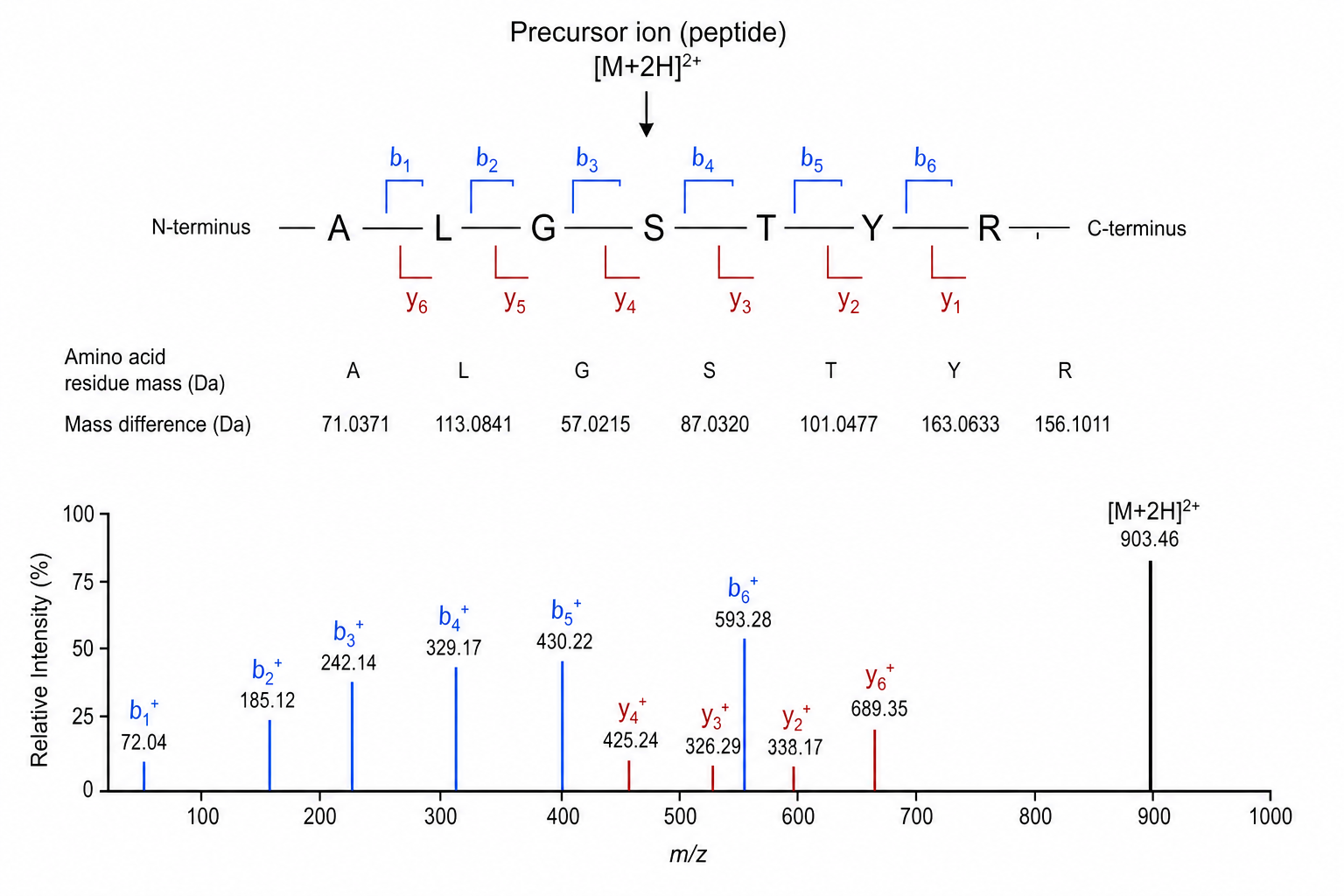
Figure 2. b-ion and y-ion series provide the core evidence used to reconstruct peptide sequence from MS/MS data.
For synthetic peptide verification, the interpreted sequence is compared directly against the target design. For unknown peptides, the assembled sequence may be used for homology searches, synthetic follow-up, or parent protein inference when additional digest data are available.
Core Advantages and Current Limitations
1. Core Advantages
(1) Direct spectrum-level evidence
Sequence calls are grounded in observed fragmentation, which is valuable for unknown peptides and independent verification of synthetic products.
(2) Compatibility with complex sample types
Purified peptides, HPLC fractions, gel-eluted bands, and digest mixtures can all support peptide-level analysis when separation and ion intensity are adequate.
(3) Flexible project scope
A project may target one critical peptide or a set of peptides from a larger digest, depending on the decision required.
(4) Support for QC and documentation
Annotated spectra and sequence reports can support batch release, publication, and tech transfer files.
2. Current Limitations
(1) Spectral quality is decisive
Weak or incomplete fragmentation limits sequence confidence, especially for long or modified peptides.
(2) Mixtures complicate interpretation
Co-eluting peptides or chimeric MS/MS spectra can produce ambiguous sequence calls if separation is insufficient.
(3) Leucine/isoleucine distinction may require additional data
Some projects accept L/I ambiguity; others require orthogonal methods for full discrimination.
(4) Single-peptide analysis does not equal full protein sequencing
Parent protein characterization may require additional digest coverage or protein-level assembly.
Typical Application Scenarios
Researchers may consider LC-MS/MS sequence analysis in the following situations:
1. Synthetic Peptide Verification
A newly synthesized peptide must be confirmed before in vivo use or customer shipment.
2. Unknown Peptide Identification
A fraction from digestion or purification contains a peptide with no reliable database match.
3. Biopharmaceutical Peptide Confirmation
A critical peptide from a biologic must be confirmed for primary structure documentation.
4. Natural Product or Bioactive Peptide Characterization
A bioactive fraction requires sequence definition before functional studies proceed.
5. Dispute Resolution After Ambiguous Database Search
A peptide-spectrum match is weak or conflicts with other evidence and requires manual sequence review.
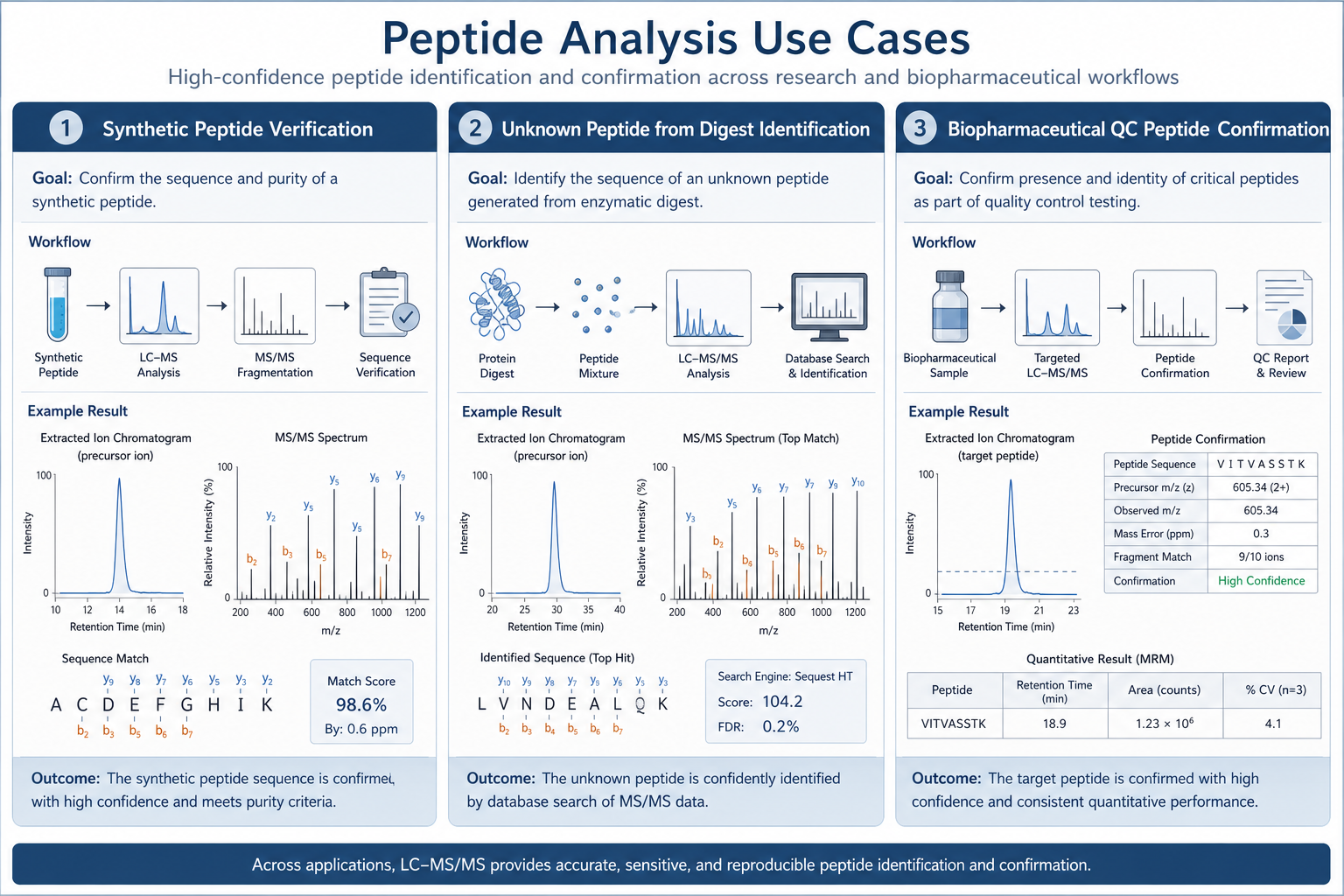
Figure 3. MS/MS-based sequencing supports synthetic verification, unknown peptide ID, and QC documentation when sequence evidence is required.
What Results Should Researchers Expect?
A strong sequencing report should include more than a sequence string. Useful deliverables often include:
Researchers should treat low-confidence regions cautiously, especially when the sequence will be used for synthesis scale-up, regulatory documentation, or parent protein inference.
Frequently Asked Questions
1. How is MS/MS peptide analysis different from protein sequencing?
The peptide-level workflow determines the sequence of one peptide from its MS/MS spectrum. Protein sequencing assembles sequence information across many peptides to define a full protein primary structure.
2. Can LC-MS/MS distinguish leucine from isoleucine?
Not always by mass spectrometry alone. Many reports note L/I ambiguity unless additional methods are applied.
3. Is database search enough for synthetic peptide verification?
Sometimes, but independent spectrum interpretation or Verification Service of Synthetic Peptide Sequence provides stronger evidence when the decision standard is high.
4. How much peptide material is typically required?
Requirements vary by purity, length, and instrument sensitivity. Feasibility review before submission is recommended.
5. Can modified peptides be sequenced?
Yes, when modification-specific mass shifts are accounted for during interpretation. Complex or multiple modifications may require extra review.
Conclusion
Peptide sequencing provides a practical route to amino acid sequence determination when MS/MS data must be interpreted directly from a peptide sample. By combining LC-MS/MS acquisition, fragmentation analysis, and expert review, the method supports synthetic verification, unknown peptide identification, and primary structure documentation at the peptide level. It is not a substitute for full protein assembly when complete primary structure is required, but it is often the fastest path when one peptide sequence is the decision point.
For synthetic verification, unknown peptide characterization, or QC documentation, MtoZ Biolabs provides Peptide Sequencing Service support covering feasibility review, LC-MS/MS analysis, spectrum annotation, and report-ready sequence deliverables. Contact the technical team to evaluate sample purity, peptide length, and the best validation path before submission.
How to order?

