





Mass Spectrometry Identify Proteins Service
Mass spectrometry identify proteins service utilizes advanced mass spectrometry techniques to determine protein molecular weight and sequence information, thereby revealing protein identity and associated post-translational modifications. In proteomics research, mass spectrometry-based protein identification is a critical technology for uncovering protein functions, elucidating cellular metabolic pathways, and discovering biomarkers. Leveraging the mass spectrometry platform, MtoZ Biolabs provides mass spectrometry identify proteins service to support solving complex scientific challenges.
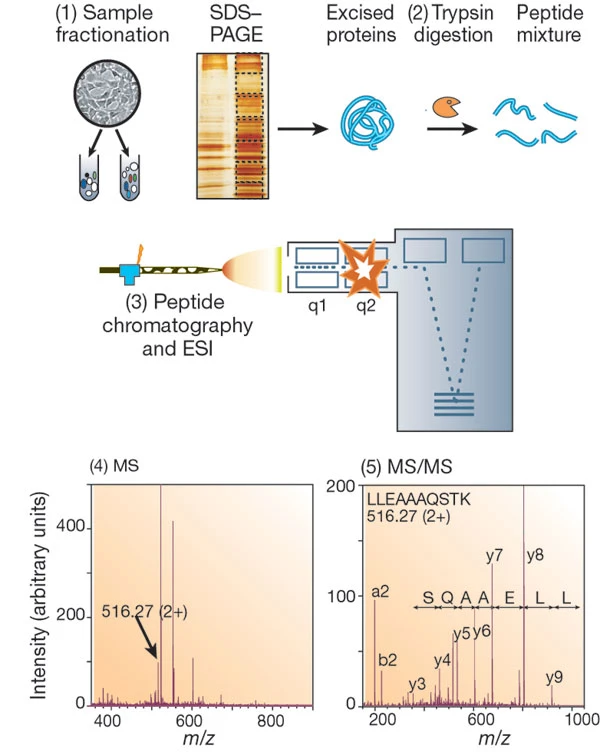
Aebersold R. et al. Nature. 2003.
Technical Principles
Mass spectrometry identify proteins service begins with enzymatic digestion of proteins into peptides. These peptides are then ionized into charged ions using either electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI) and analyzed based on their mass-to-charge ratio (m/z). In primary mass spectrometry (MS1), the molecular weights of individual peptides are measured, generating a peptide mass spectrum. In secondary mass spectrometry (MS2), specific peptide ions are selectively fragmented, and the masses of the resulting fragment ions are measured, producing a fragment spectrum. This fragment spectrum is then used to deduce the amino acid sequence of the peptide. The mass spectrometry data are compared against protein databases to match known peptide sequences. Using these peptides, researchers can infer protein identity, sequence coverage, and post-translational modifications (e.g., phosphorylation, glycosylation).
Service Advantages
1. Advanced Analysis Platform: MtoZ Biolabs established an advanced mass spectrometry identify proteins service platform, guaranteeing reliable, fast, and highly accurate analysis service.
2. One-Time-Charge: Our pricing is transparent, no hidden fees or additional costs.
3. High-Data-Quality: Deep data coverage with strict data quality control. AI-powered bioinformatics platform integrates all mass spectrometry identify proteins service data, providing clients with a comprehensive data report.
4. Various Sample Types Supported: MtoZ Biolabs offers protein identification services for a wide range of sample types, including purified samples, protein gel spots, protein gel bands, pull-down samples, co-immunoprecipitation (Co-IP) samples, and complex samples, ensuring versatility to meet diverse research needs.
Applications
The mass spectrometry identify proteins service is widely utilized in the following research areas:
Proteomics Research
Systematic analysis of the types and expression levels of proteins in samples. Comprehensive identification and quantification of proteins to uncover dynamic changes in cellular or tissue proteomes.
Post-Translational Modification Analysis
Efficient detection of modifications such as phosphorylation and glycosylation to elucidate their roles in regulating protein functions.
Biomarker Discovery
Through differential protein identification, potential disease-related markers are discovered for diagnosis and prognosis assessment.
Protein Interaction Analysis
Mass spectrometry combined with affinity capture techniques (e.g., Pull-Down, Co-IP) is used to study protein-protein, protein-nucleic acid, or protein-small molecule interactions. This includes analyzing drug-target interactions to optimize drug design and validate therapeutic effects.
Novel and Mutant Protein Identification
Facilitates the identification of novel proteins or proteins with sequence variations resulting from gene mutations.
FAQ
Q: How to Enhance Detection Efficiency for Low-Abundance and Membrane Proteins?
Detection of low-abundance and membrane proteins is often challenged by low signal intensity and high sample loss. The following strategies can effectively enhance detection efficiency:
1. Sample Enrichment
Low-Abundance Proteins: Use affinity capture, immunoenrichment, or surface-enhanced techniques (e.g., SP3 magnetic beads) to enrich target proteins.
Membrane Proteins: Apply detergents (e.g., CHAPS or Triton X-100) or non-ionic surfactants to extract membrane proteins, followed by enrichment using membrane protein-specific columns.
2. Optimize Sample Separation
Utilize high-performance liquid chromatography (e.g., high-pH reversed-phase separation) or electrophoresis to reduce sample complexity and improve the separation efficiency of peptides derived from low-abundance or membrane proteins.
3. Adjust Mass Spectrometry Parameters
Increase Sensitivity: Optimize dynamic exclusion times to prioritize the collection of low-abundance peptides.
Select Appropriate Fragmentation Modes: Employ fragmentation methods such as HCD (Higher-Energy Collisional Dissociation) or ETD (Electron Transfer Dissociation), which are more suitable for low-abundance peptides.
4. Isotopic Labeling or Tag-Based Quantification
Enhance the reliability of low-abundance signals using quantification technologies like Tandem Mass Tag (TMT).
5. Deep Sample Acquisition
Extend gradient elution times to increase resolution in liquid-phase separation, enabling more comprehensive protein coverage.
By implementing these strategies, the detection efficiency of low-abundance and membrane proteins can be significantly improved, supporting the identification of critical proteins in low-intensity signals or complex samples.
Case Study
Using mass spectrometry identify proteins researchers identified multiple phosphorylation sites of Cdc25C, a critical cell cycle regulatory protein. Further investigation into the regulatory effects of phosphorylation on Cdc25C activity revealed that specific phosphorylation modifications play a pivotal role during M-phase induction by regulating Cdc25C activity, thereby facilitating the proper progression of the cell cycle.
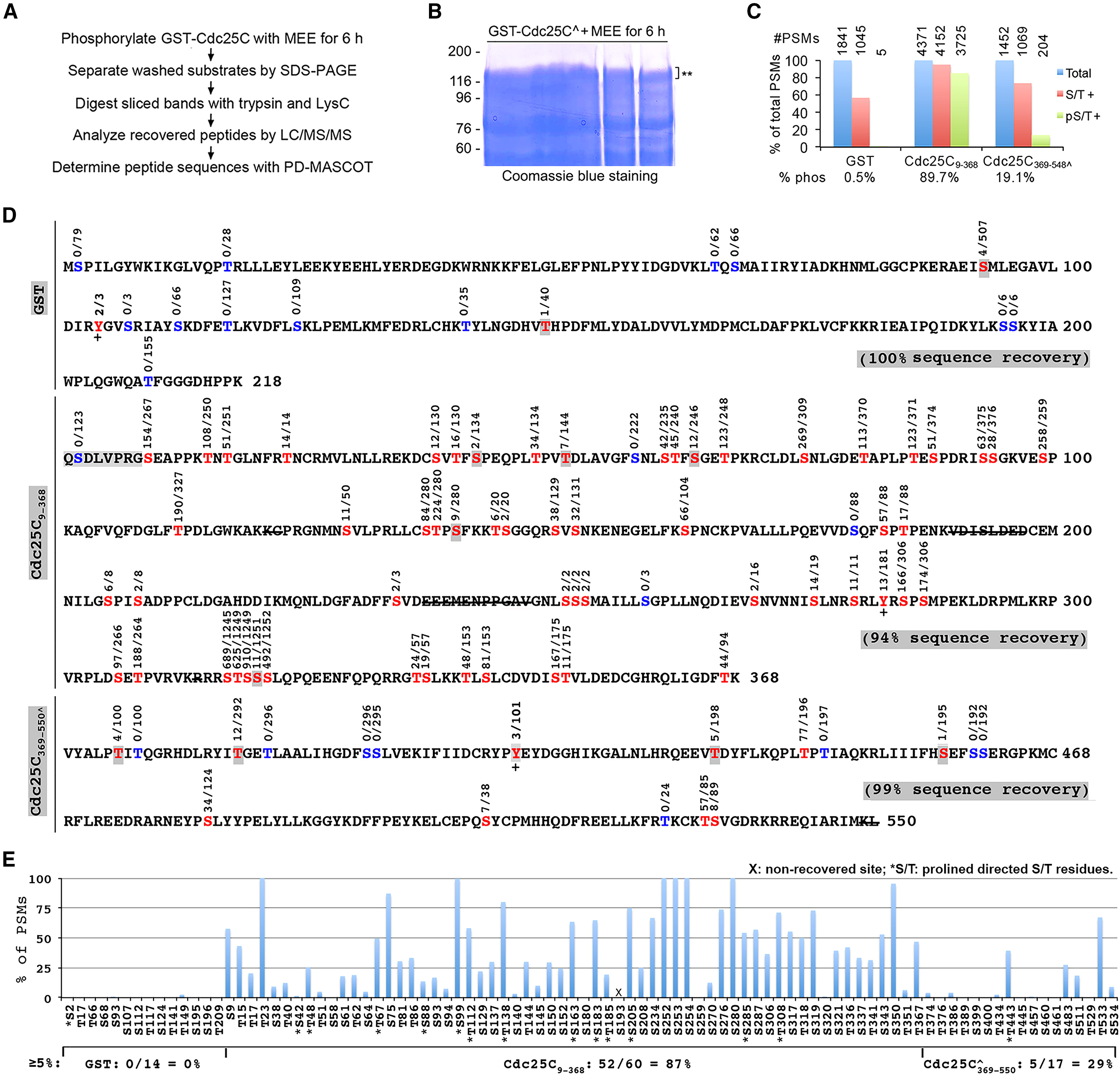
Tan T. et al. iScience. 2025.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Unknown Protein Identification Service
How to order?

