





Interpreting PhIP-Seq Results: Enriched Peptides, Cross-Reactivity, Controls, and Biological Relevance
An enriched peptide in a PhIP-Seq report is only potentially meaningful after three checks are cleared, in order: the peptide-level enrichment is credible against controls, the signal fits a coherent antigen context, and the interpretation still stands after review for shared-motif or homology-driven cross-reactivity. If a hit appears only as an isolated peptide, overlaps with background binding in mock IP or bead-only control samples, or is driven by a short motif that recurs across unrelated proteins, it is usually a weak basis for downstream claims.
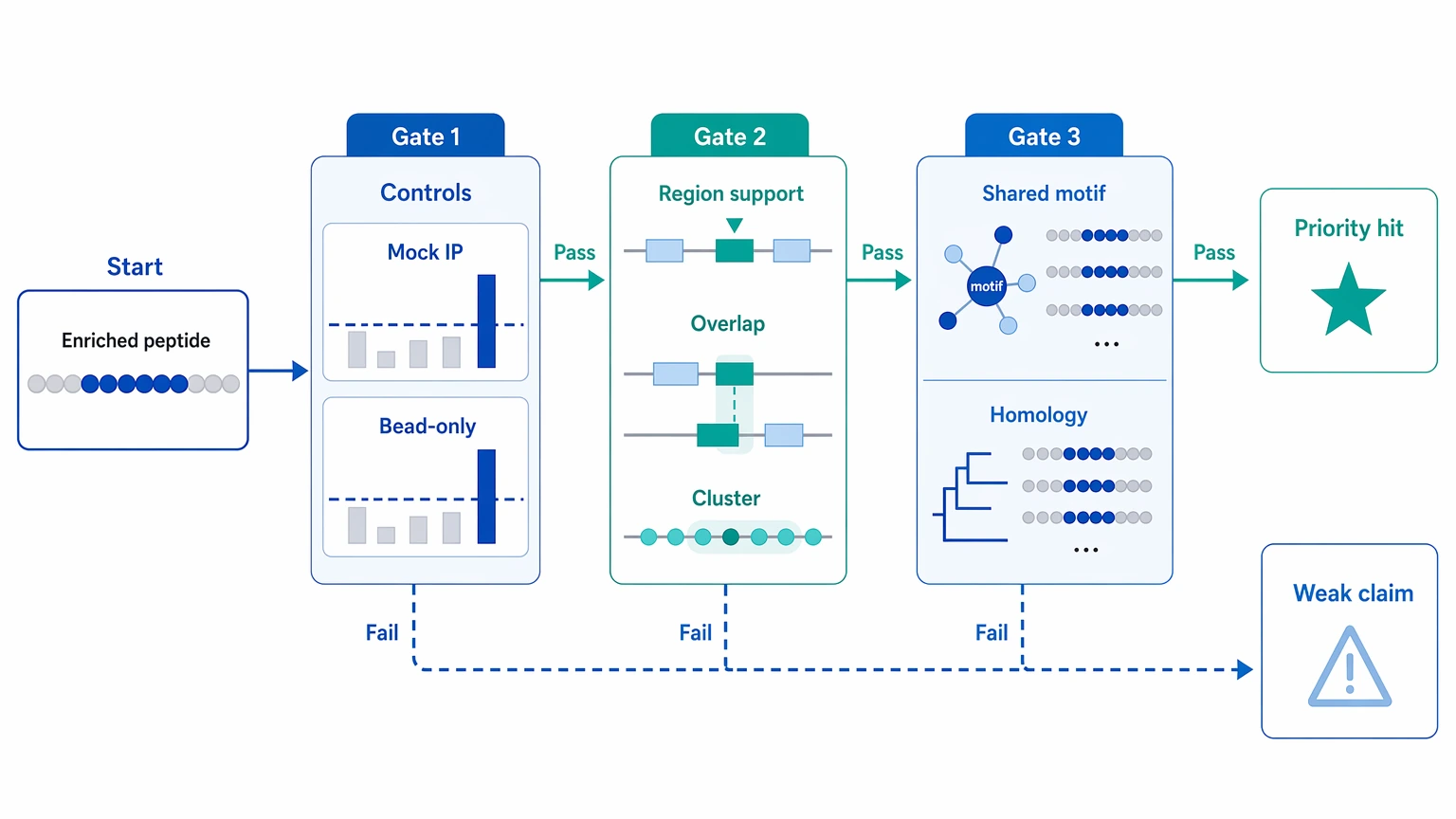
For practical phip seq analysis, start with normalized read counts, fold enrichment, a model-based enrichment statistic such as a z-score, and the adjusted p-value or false discovery rate. Then ask whether replicate concordance, peptide tiling, and cohort comparison point in the same direction. Only after that should you decide whether a peptide belongs on a shortlist for peptide ELISA, a competition assay, alternative serology, or antigen-focused follow-up.
Where interpretation usually becomes difficult
The hard part often starts after primary enrichment calling is done. A team gets a report with dozens or hundreds of enriched peptides. Some cluster within one protein region, which looks encouraging. Others also show up in negative controls at lower levels. A few top-ranked hits map to short shared motifs across several taxa or unrelated proteins, and that makes the biological meaning hard to pin down.
At that stage, the question is no longer which peptides are enriched, but which enrichments deserve trust. That choice affects hit prioritization, cohort comparison claims, and whether the next round of work should focus on validation or redesign. Weak hits pushed forward too early can consume sample and budget. Strong clustered signals written off as noise can hide the best linear epitope candidates in the dataset.
The main sources of ambiguous PhIP-Seq signals
Background binding visible in controls
Some peptides enrich because they bind nonspecifically during immunoprecipitation or interact with beads, capture reagents, or recurrent phage features. When the same peptides recur in mock IP, bead-only control, blank, or negative control samples, they may be recurrent background binders rather than sample-specific antibody recognition.
Isolated hits with limited antigen context
A single enriched peptide can be statistically significant and still stay biologically ambiguous. Without support from overlapping peptides, positional clustering, or repeat detection in related samples, it is harder to read that hit as a credible linear epitope signal.
Cross-reactivity driven by a shared motif or sequence homology
Short motifs can create apparent reactivity across unrelated proteins or organisms. Sequence homology can do the same across members of the same protein family. These hits are not automatically false, but they often support a motif-level interpretation better than a source-antigen claim.
Overcalled cohort comparison
Case-control differences can look stronger than they really are when prevalence is low, sample batches are imbalanced, or many peptides are tested with sparse counts. Even a passing false discovery rate threshold does not establish biological relevance by itself.
Related Services
Main Service
Supporting Service
A step-by-step framework for phip seq analysis and interpretation
Step 1: Confirm that peptide-level enrichment is technically credible
Start with raw and normalized read counts. Review whether the peptide has enough baseline representation in the phage-displayed peptide library to support stable interpretation, then compare fold enrichment against input, mock IP, or another relevant baseline. A model-based enrichment statistic, such as a z-score or negative binomial score, should broadly agree with the ranking rather than point somewhere else. The adjusted p-value helps control multiple testing, but it should not be the only filter.
A technically credible hit usually stays notable across more than one metric. It should not owe its position entirely to count sparsity, unstable denominators, or extreme values produced by a poorly represented peptide.
Step 2: Read the controls before trusting the top-ranked hits
Controls are often the fastest way to weaken an overconfident interpretation. Check whether the same peptide, motif class, or peptide family appears in mock IP, bead-only control, blank, or negative control samples. Then compare both the magnitude and frequency of those control signals with the study samples. A peptide that appears broadly in controls may still enrich more strongly in a subset of samples, but the claim becomes narrower: it is a candidate rising above a known background class, not a clean sample-specific discovery.
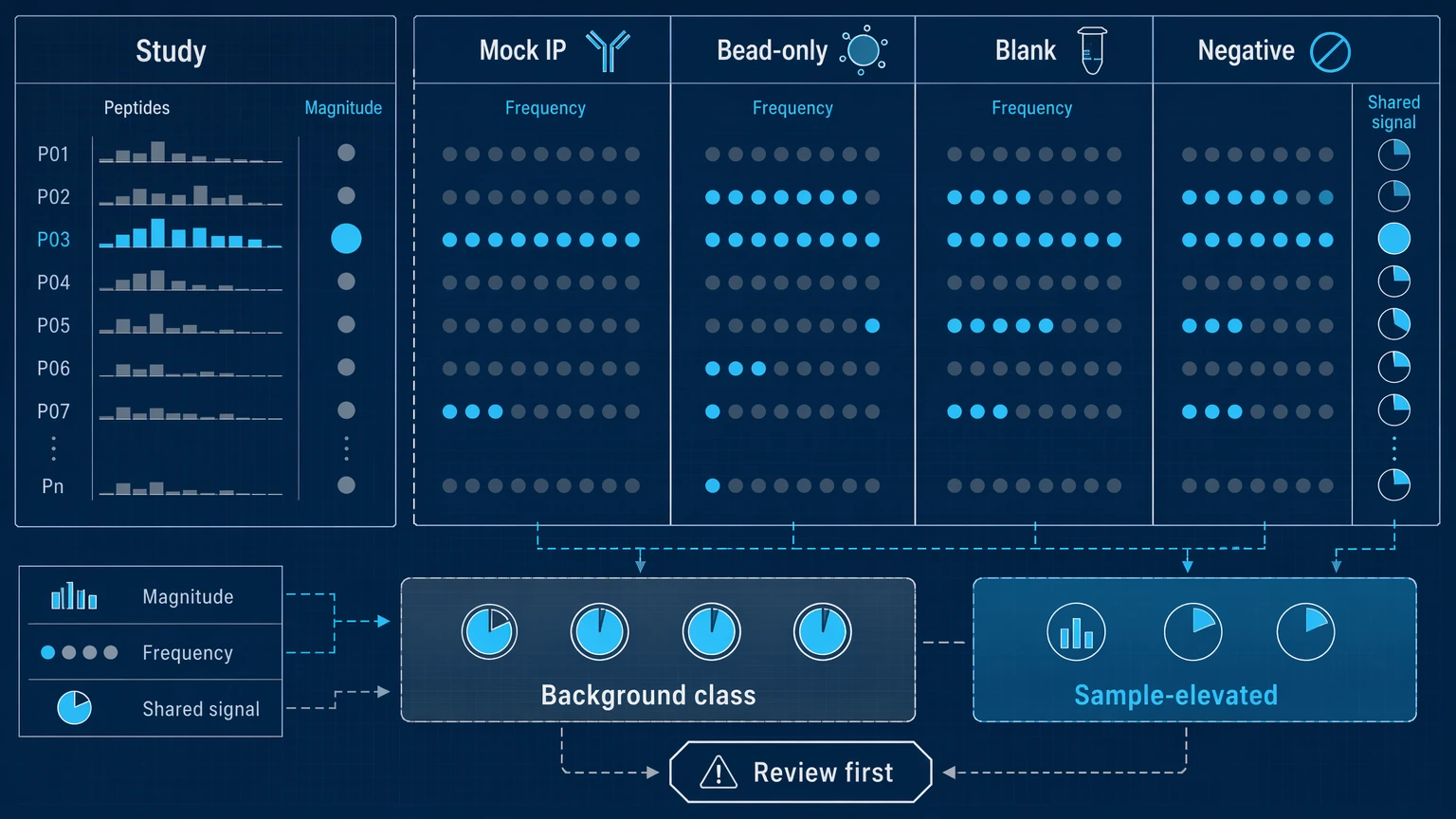
Replicate concordance matters here too. If technical repeats disagree on rank, direction, or enrichment status, that peptide should not move forward as a high-confidence hit. When control structure is complicated or the report language does not match the data pattern, you can submit your requirements to MtoZ Biolabs to evaluate your project in the context of PhIP-Seq controls, analysis thresholds, and report interpretation.
Step 3: Decide whether the pattern supports a coherent antigen region
The strongest PhIP-Seq interpretations usually come from peptide tiling, not from one isolated sequence. Look for overlapping peptides enriched within the same region of the source protein. A cluster of adjacent hits suggests repeated sampling of the same linear epitope neighborhood, which is much harder to explain by random pull-down alone.
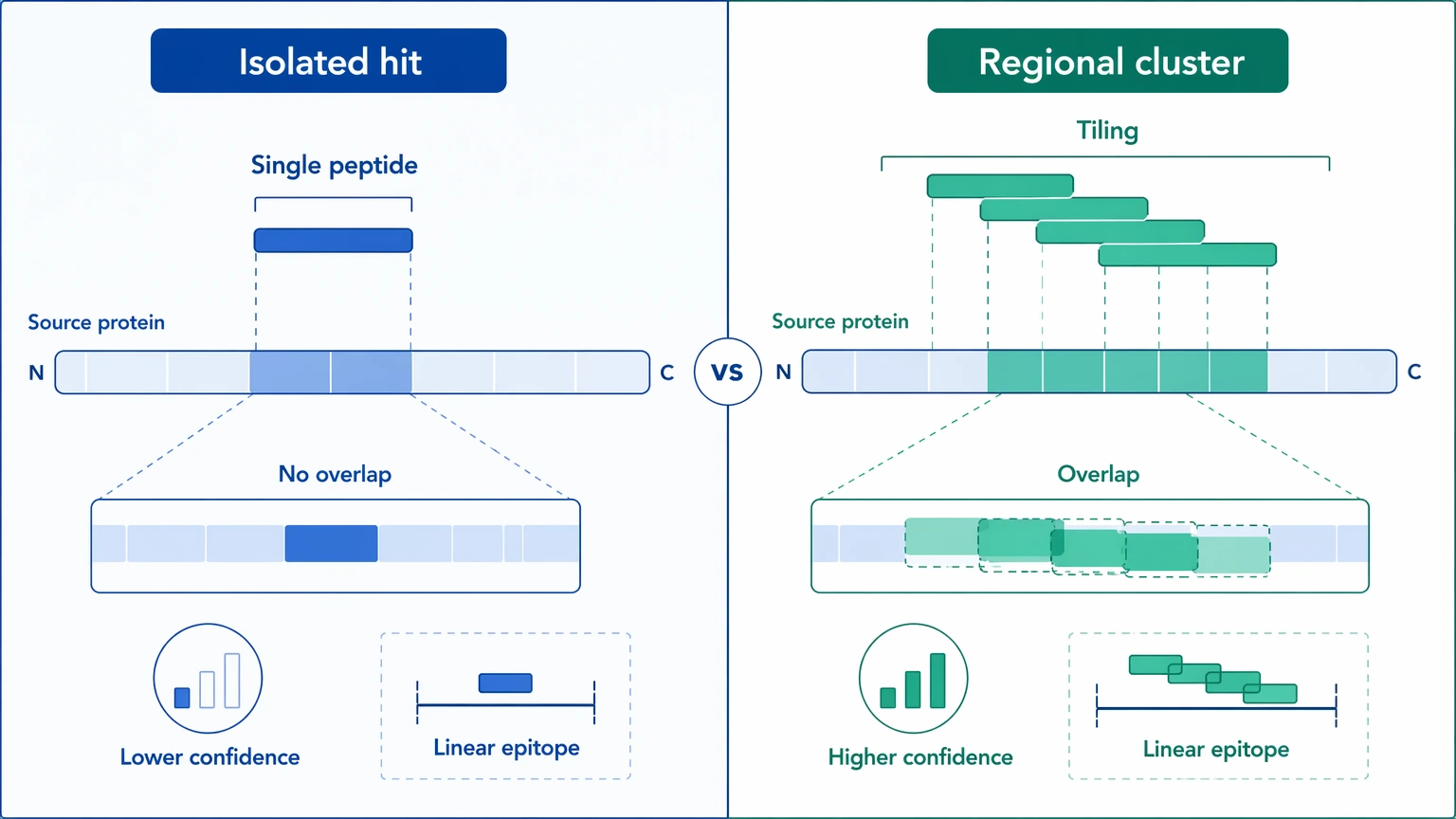
Antigen context matters as much as peptide count. Ask whether the enriched region is plausible in the source protein and whether several samples show the same local pattern. Multi-peptide regional support does not prove function or specificity, but it does raise confidence that the signal reflects reproducible antibody recognition. By contrast, one peptide with no neighboring support often belongs on a watch list rather than a validation list.
Step 4: Test for cross-reactivity instead of assuming specificity
Next, examine whether the enriched peptide shares a short motif with hits from unrelated proteins or taxa. Shared motif analysis helps separate antigen-specific interpretation from motif-driven cross-reactivity. Sequence homology review serves a similar role across related proteins. In many datasets, the antibody-binding event is real at the peptide level, but the antigen assignment stays ambiguous.
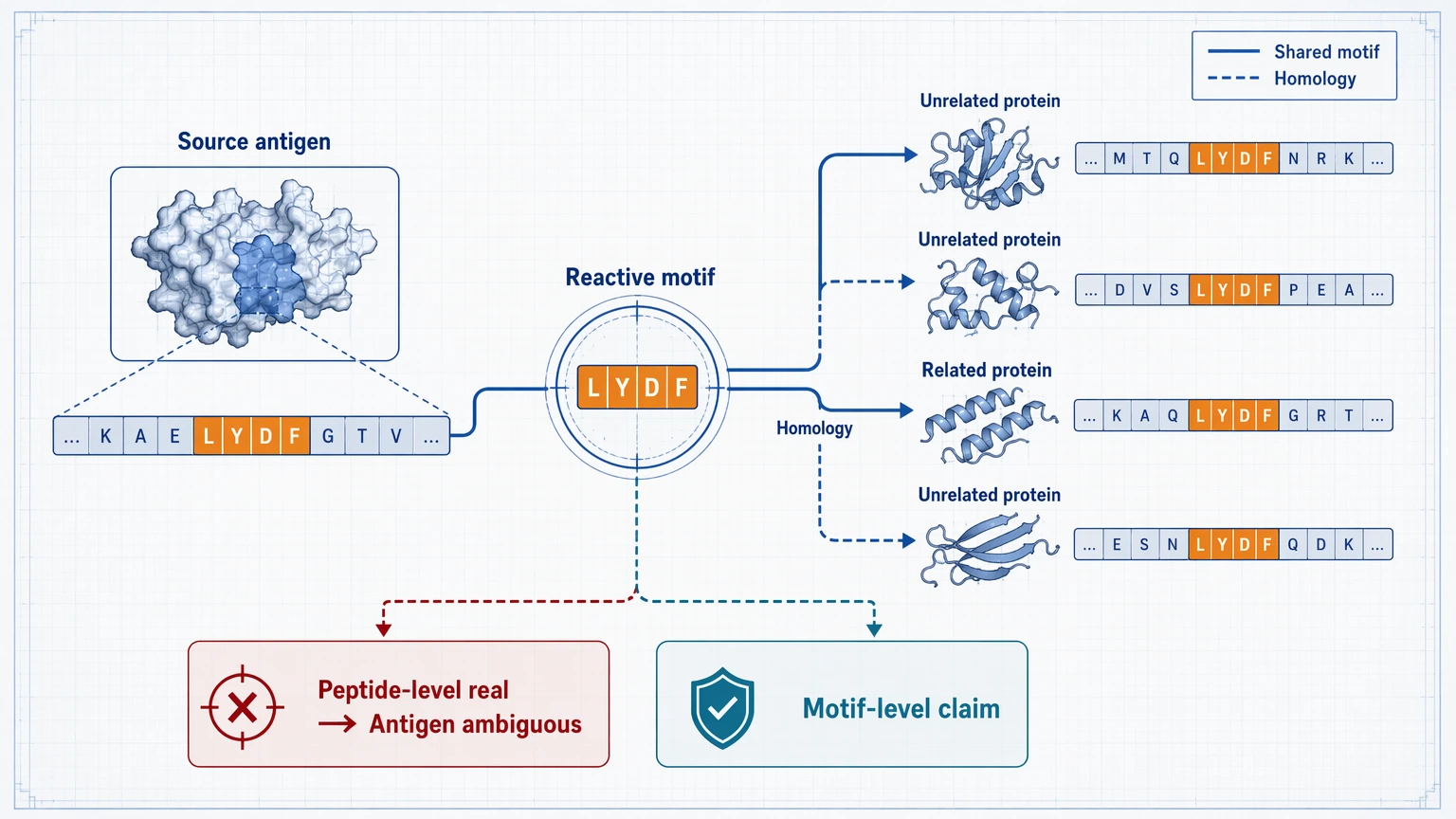
That distinction changes how results should be written. If several proteins carry the same reactive motif, the most defensible statement may be that antibodies recognize a motif class rather than one specific source antigen. For mechanistic immunology, that can still be informative. For biomarker screening, though, such hits usually drop on the priority list unless orthogonal validation can sort out specificity.
Step 5: Review cohort comparison with effect size, prevalence, and batch stability
For cohort comparison, ask three questions: how many samples carry the hit, how large the between-group shift is in normalized read counts or enrichment score, and whether the pattern holds across batches, sequencing runs, or subcohorts. A peptide with a strong adjusted p-value but very low prevalence may not support a stable group-level conclusion. Likewise, broad but weak enrichment can produce nominal significance without much biological weight.
Useful outputs at this stage include prevalence by cohort, distributional differences in normalized read counts, consistency across technical batches, and recurrence of the same antigen region in independent subsets. If the cohort signal disappears after batch-aware review, the right next step is to revise the comparison strategy rather than escalate the claim.
Step 6: Build a validation-ready shortlist
A validation-ready candidate usually shows most of the following: credible peptide-level enrichment, low background binding in controls, acceptable replicate concordance, positional support from overlapping peptides, interpretable antigen context, and limited ambiguity from shared motifs or sequence homology. Cohort-linked candidates should also show a clear prevalence or effect-size pattern that stays stable after batch review.
The follow-up assay should match the interpretation question. Peptide ELISA is useful when you want to test whether the same linear epitope signal reproduces in a targeted format. A competition assay is useful when you need to ask whether nearby peptides or homologous sequences are capturing the same antibody population. Protein-based or alternative serology assays become more informative when the peptide pattern suggests broader antigen-level reactivity than a single linear epitope claim can support.
Expected results and how to verify them
After this framework is applied, the immediate result should be a smaller, cleaner list of candidates. Some peptides will be downgraded to background binding, recurrent background binders, or shared-motif cross-reactive signals. Others will move up because they show stronger replicate concordance, lower control overlap, and better antigen context than their raw ranks first suggested.
Verification should be tied to observable outputs. Short-term checks include stable ranking across replicate samples, cleaner separation from mock IP or bead-only controls, recovery of overlapping peptides in the same antigen region, and more consistent cohort prevalence after batch-aware review. Longer-term confirmation may require peptide ELISA, a competition assay, or a protein-based assay to test whether the shortlisted signal persists in an orthogonal format.
The reporting language should get cleaner too. Instead of saying that a peptide is positive, the report can state whether it represents a control-resilient enriched peptide, a clustered linear epitope candidate, a likely shared motif signal, or a cohort-associated hit that still needs orthogonal validation. That narrower wording is more accurate and more useful for downstream planning.
Key caution points before moving into validation
Interpretation boundaries should match assay format. PhIP-Seq measures enrichment of peptides captured after immunoprecipitation from a phage-displayed peptide library, so it primarily informs linear epitope reactivity. It does not directly establish conformational epitope binding to native proteins, clinical diagnosis, or calibrated quantitative equivalence to targeted immunoassays.
Review sample quality and sample amount before any follow-up. Limited serum or plasma volume can restrict repeat testing, competition assays, or platform switching. If sample input is low, prioritize the most control-resilient and region-supported candidates first instead of expanding the shortlist too early.
Controls and repeats should be planned before interpretation is finalized. Missing mock IP, bead-only, blank, or well-matched negative controls make it harder to separate sample-specific enrichment from recurrent background binding. Technical repeats and balanced cohort design add cost and time, but without them, prevalence claims and batch-sensitive differences remain harder to defend.
Batch structure and contamination risk also need direct review. If cases and controls were processed in different runs, or if reagent lots changed during library preparation or immunoprecipitation, group differences should be treated conservatively until the signal survives batch-aware analysis. Recurrent contaminants and sticky peptide classes should be tracked across runs instead of being judged one sample at a time.
Finally, know when to change methods or seek external review. If the main question depends on native conformation, close homolog discrimination, or functional antibody behavior, a protein-based assay or alternative serology format may be more appropriate than further peptide-level interpretation. If your dataset sits between background binding and a validation decision, contact MtoZ Biolabs to discuss the study, evaluate the project, and plan a follow-up path that fits the sample set, controls, and report structure.
Conclusion
Strong PhIP-Seq interpretation comes from tying statistics to context: confirm peptide-level enrichment with normalized read counts, fold enrichment, and model-based significance; challenge the signal with mock IP, bead-only control, and negative control behavior; look for replicate concordance and overlapping peptides within a coherent antigen region; and then test whether cross-reactivity, shared motifs, or sequence homology explain the pattern better than a single-antigen claim. This approach fits antibody profiling, biomarker screening, and translational immunology projects that need a defensible shortlist for orthogonal validation rather than an expanded list of nominal hits.
FAQ
Can cross-reactive hits still be useful, or should they be removed from the report?
They can still be useful. A cross-reactive signal may indicate a shared motif, a conserved protein family feature, or broader immune recognition. The key is to label it correctly and avoid overstating antigen specificity before orthogonal validation.
What should you do if the top hits look convincing but the control set is limited?
Treat the interpretation as provisional. You can still review replicate concordance, peptide clustering, and motif overlap, but missing mock IP or bead-only control data should lower confidence and may justify adding controls before major follow-up work.
When should you move from peptide ELISA to a protein-based assay?
Move to a protein-based assay when the scientific question depends on full antigen context, discrimination among homologous proteins, or whether the peptide signal translates to broader antigen recognition. Peptide ELISA is more directly aligned with linear epitope confirmation.
Do longitudinal samples change how you prioritize hits?
Yes. For longitudinal studies, persistence, gain, or loss of the same enriched region over time can be more informative than a single timepoint rank. Stable recurrence across timepoints usually carries more weight than one isolated peak.
How should uncertain findings be described in a report?
Use bounded language. For example, describe a peptide as a candidate enriched peptide with control-limited specificity or a shared-motif cross-reactive signal requiring orthogonal validation, rather than presenting it as a confirmed antigen-specific antibody response.
How to order?

