





How to Use SWATH-MS for Label-Free Protein Quantification?
In proteomics research, label-free quantification (LFQ) has become an increasingly prominent approach for differential protein profiling and mechanistic studies. This is largely attributed to its advantages, including the elimination of costly labeling reagents, scalability with respect to sample number, and compatibility with clinical specimens. Among the diverse LFQ strategies, SWATH-MS—representing a data-independent acquisition (DIA)-based method—integrates the broad identification capability of data-dependent acquisition (DDA) with the quantitative consistency of MS1-based approaches. This makes SWATH-MS particularly well-suited for high-throughput studies requiring robust reproducibility. As a prototypical DIA technique, SWATH-MS not only addresses the issues of missing values and low reproducibility inherent in traditional DDA workflows but also enables higher throughput and expands the dynamic range of protein quantification. This article provides a systematic overview of how to employ SWATH-MS for label-free quantification, covering its fundamental principles, experimental workflow, data analysis strategies, and practical applications to fully elucidate its technical utility.
Principle of SWATH-MS: Broader Coverage, Greater Quantitative Reliability
The central strength of SWATH-MS lies in segmenting the entire m/z scan range into dozens of discrete acquisition windows, within which all precursor ions are fragmented and their corresponding fragment ion spectra recorded in a comprehensive and systematic manner. This approach eliminates the high-abundance peptide bias typically observed in DDA methods and produces datasets with strong quantitative reproducibility and suitability for retrospective analysis. Unlike MS1-based LFQ, which quantifies peptides based on precursor ion intensities, SWATH-MS performs quantification at the MS2 level using fragment ions, thereby offering enhanced selectivity and resistance to interference from co-eluting species.
Standard Workflow for SWATH-MS-Based Label-Free Quantitative Analysis
The application of SWATH-MS for label-free protein quantification involves four critical steps, each of which significantly influences the quality and interpretability of the final data.
1. Protein Extraction and Digestion
Efficient and consistent protein extraction is essential for ensuring reliable mass spectrometry performance in downstream analysis. The commonly adopted protocol includes:
(1) Lysing samples using buffer solutions containing denaturing agents (e.g., 8 M urea or 4% SDS)
(2) Performing DTT-mediated reduction followed by IAA alkylation to stabilize disulfide bonds
(3) Enzymatic digestion with trypsin, typically carried out for 12–16 hours
MtoZ Biolabs employs a magnetic bead-based micro-purification approach combined with a temperature-controlled automated digestion platform, which significantly enhances digestion efficiency and reproducibility. This setup is particularly beneficial for SWATH-MS workflows, where peptide uniformity is critical for accurate quantification.
2. LC-MS Acquisition Settings
Chromatographic and mass spectrometric configurations are pivotal in determining the depth and precision of SWATH-MS data acquisition. A typical setup includes:
(1) LC system: nano-flow liquid chromatography (e.g., EASY-nLC 1200) with a gradient duration of 60–120 minutes
(2) MS platforms: Sciex TripleTOF, Thermo Exploris, or Bruker timsTOF
(3) SWATH acquisition range: 400–1200 m/z
(4) Number of SWATH windows: 64–100 (with optional overlap), each window approximately 25 Da wide
Finer window segmentation provides higher resolution but also increases acquisition time. Therefore, the window design must be optimized based on the complexity of the sample to achieve a balanced trade-off between resolution and throughput.
3. Spectral Library Construction and Matching Strategies
The spectral library is a core component of SWATH-MS-based identification, containing peptide fragmentation patterns acquired through mass spectrometry. The construction approaches include:
(1) Experimental spectral libraries: generated from data-dependent acquisition (DDA) of the same sample batch, offering high peptide coverage and high accuracy.
(2) Public spectral libraries: such as Pan-Human and SWATHAtlas, which are applicable to well-characterized model organisms.
(3) Predicted spectral libraries: tools like Prosit and DeepMass employ artificial intelligence to predict peptide fragmentation spectra, suitable for newly studied species or cases with limited sample availability.
MtoZ Biolabs can integrate multiple types of spectral libraries based on project goals to enhance peptide identification rates while balancing throughput and cost-effectiveness.
4. DIA Data Analysis
Recommended Analytical Tools (Selected Based on Budget and Required Technical Expertise):

Typical data processing steps include:
(1) Matching peptides against the spectral library
(2) Multidimensional scoring and false discovery rate (FDR) control
(3) Retention time (RT) alignment
(4) Exporting protein quantification matrices based on intensity or peak area
According to client requirements, MtoZ Biolabs provides protein quantification matrices, differential expression analyses, functional enrichment (GO/KEGG) reports, and visualization charts to support timely and efficient scientific publication.
Comparison Among SWATH-MS and Other Quantification Methods
When selecting a protein quantification strategy, researchers are often confronted with several methodological choices, including DDA-LFQ, TMT/iTRAQ labeling approaches, and SWATH-MS. A representative comparison is summarized below:
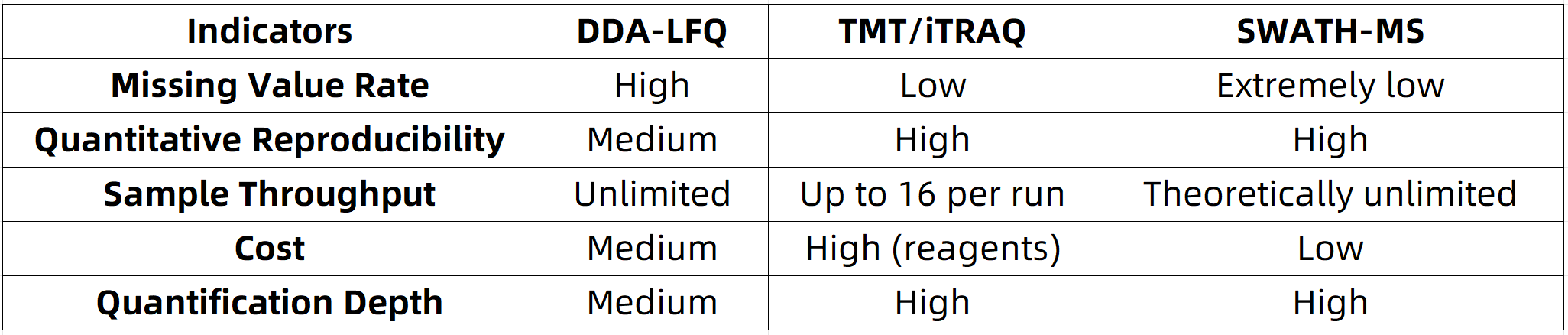
SWATH-MS Solutions by MtoZ Biolabs
MtoZ Biolabs has supported hundreds of SWATH-MS projects, offering the following key advantages:
1. Multi-platform compatibility: Orbitrap Exploris 480 / timsTOF Pro / TripleTOF
2. Broad sample type coverage: tissues, serum, cells, exosomes, FFPE, and more
3. Flexible spectral library strategy: integrated use of public libraries, experimental libraries, and AI-predicted libraries
4. High-quality deliverables: charts and data conforming to publication standards in SCI-indexed journals
SWATH-MS is increasingly recognized as a new standard in modern proteomics research, particularly advantageous in studies involving large sample sizes, constrained budgets, and strict data reproducibility requirements. By leveraging AI-powered spectral libraries, advanced computational algorithms, and high-performance mass spectrometry platforms, SWATH-MS is poised to expand beyond academic research into applications in disease diagnostics, drug discovery, and precision medicine. MtoZ Biolabs empowers researchers to conduct high-throughput proteomic investigations with greater efficiency and cost-effectiveness.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

