





How to Optimize PhIP-Seq Immunoprecipitation Efficiency?
Introduction
Low immunoprecipitation efficiency is one of the most common reasons a PhIP-Seq experiment produces weak, noisy, or difficult-to-interpret results. A serum sample may contain meaningful antibodies, but the final sequencing data can still show poor peptide enrichment when antibody binding, bead capture, washing, or library representation is not controlled. In autoimmune disease, infectious disease serology, vaccine response, or biomarker discovery, this can turn a promising sample set into an ambiguous hit list.
PhIP-Seq immunoprecipitation efficiency is not determined by one reagent or one step—it reflects the combined performance of sample quality, peptide library balance, antibody-library incubation, capture chemistry, wash stringency, sequencing depth, and data filtering. Optimizing only one variable without checking the others may improve signal in one area while increasing background in another.
A practical optimization strategy should begin with the experimental question. Broad serum antibody profiling must preserve library diversity while enriching true antibody-bound clones. Focused epitope discovery needs enough stringency to separate specific binders from carryover. If your team needs to identify whether low recovery is driven by samples, controls, or capture conditions, MtoZ Biolabs can review the PhIP-Seq workflow before the cohort is consumed.
Related Services
|
Research Need |
Recommended Service |
|---|---|
|
Need broad antibody reactivity profiling from serum or plasma |
|
|
Need peptide-level epitope discovery after screening |
|
|
Need targeted validation of candidate peptide regions |
|
|
Need broader peptide epitope screening support |
Why PhIP-Seq Immunoprecipitation Efficiency Drops
The first step in troubleshooting PhIP-Seq is to define what "low efficiency" means in the dataset. It may appear as low recovered DNA, poor sequencing library yield, weak enrichment over controls, high background binding, loss of expected positive controls, or poor replicate overlap. These problems may look similar at the end of the workflow, but their causes can be different.
Weak antibody signal is one possible cause. Serum antibodies may be low abundance, degraded, poorly matched to the peptide library, or affected by sample handling. If the relevant antibodies are rare, too little input or overly harsh washes can push real binders below the detection threshold.
Library imbalance is another frequent issue. A phage display peptide library should maintain sufficient diversity throughout the experiment. If library input is too low, a bottleneck can occur before antibody binding is measured. If the library is degraded or unevenly amplified, sequencing reads may reflect library structure rather than true enrichment.
Capture-related problems can also reduce recovery. Magnetic beads, capture proteins, incubation time, and bead-to-antibody ratio all affect the final pull-down. Too little bead capacity can miss antibody-bound phage. Too much bead material can raise nonspecific pull-down and increase background.

Figure 1. Low IP efficiency can come from weak antibody signal, library bottlenecks, bead background, or overly harsh washes.
Step 1: Start with Sample Quality and Input Consistency
Sample quality should be checked before changing the IP protocol. Serum and plasma samples are sensitive to collection method, storage condition, freeze-thaw history, hemolysis, lipid content, and contamination. Inconsistent sample handling can produce batch effects that look like biological differences.
Researchers should record sample type, collection tube, storage temperature, freeze-thaw cycles, and time in storage. Samples from different groups should be processed in a balanced order so technical variation is not mistaken for antibody enrichment.
Input normalization is also important. Inconsistent serum volumes or antibody concentrations can distort group comparison. In discovery studies, use a consistent input strategy across the cohort and rely on controls to interpret sample-to-sample variation.
Step 2: Preserve Peptide Library Diversity
A PhIP-Seq library is informative only if the relevant peptide clones are adequately represented. Before optimization, researchers should check library quality, titer, diversity, and baseline distribution. A poorly balanced library can create false negatives because some peptides are underrepresented before the pull-down begins.
Library input should be high enough to avoid bottlenecking. If too few phage particles enter the binding reaction, stochastic sampling can dominate the result, especially in broad peptidomes, pathogen panels, or large custom libraries.
More library is not always better. Excessive input can increase nonspecific interactions and make true enrichment harder to detect. A useful pilot tests a limited range of inputs while monitoring recovered DNA, control enrichment, and replicate consistency.
Step 3: Optimize Antibody-Library Binding Conditions
Antibody binding depends on incubation time, temperature, mixing, buffer composition, and serum dilution. Short incubation may reduce recovery of lower-affinity binders. Very long incubation can increase nonspecific interactions. The best condition improves specific enrichment without raising background.
Serum dilution is a key variable. Concentrated serum may increase nonspecific binding, matrix effects, or antibody competition. Over-diluted serum may reduce low-abundance antibody recovery. A small dilution series can show whether the assay is limited by antibody concentration or background noise.
Buffer composition should support antibody-antigen interaction while reducing nonspecific adhesion. Salt concentration, detergent level, blocking reagent, and pH can all affect signal and background. Change these conditions one at a time whenever possible, because simultaneous changes make troubleshooting difficult.
Step 4: Balance Bead Capacity and Nonspecific Capture
Magnetic bead capture is often where PhIP-Seq signal is either preserved or diluted by background. Bead type, bead amount, capture protein, blocking strategy, and wash compatibility should match the antibody class, subclass, and sample type.
Too little bead capacity can reduce recovery of antibody-bound phage. Too much bead material can increase nonspecific adsorption and carryover. The goal is selective recovery of antibody-bound peptide clones with acceptable background.
Blocking conditions can reduce nonspecific binding, but excessive blocking may interfere with capture. Compare negative controls, no-serum controls, and known positive samples when adjusting bead conditions. A change is useful only if it improves specific enrichment relative to background.
Step 5: Adjust Wash Stringency Carefully
Wash steps remove nonspecific material, but they can also remove real antibody-peptide interactions. Mild washes leave background. Harsh washes may lose weak but meaningful binders, especially in discovery projects with varied antibody affinity.
Wash optimization should consider wash number, volume, salt, detergent, mixing intensity, and time per wash. Compare a standard condition with one milder and one stricter condition. Judge the result by enrichment quality, control behavior, and replicate overlap.
Avoid using recovered DNA amount as the only success metric. High recovery may simply reflect nonspecific pull-down. A lower DNA yield with stronger enrichment and cleaner controls may be more useful.
Step 6: Use Controls to Separate Signal from Background
Controls are essential for interpreting IP results. A no-serum control helps measure nonspecific phage or bead binding. A healthy control group can define background reactivity in cohort studies. Technical replicates evaluate reproducibility. Positive controls, when available, confirm that the assay can recover expected antibody-peptide interactions.
The most useful controls are planned before the experiment begins. Adding controls after poor data are observed may help diagnosis, but it cannot fully rescue a weak study design. For disease-control or treatment-response projects, process controls alongside experimental samples.
For limited serum volume or cohort comparisons, MtoZ Biolabs can help design a PhIP-Seq workflow with appropriate sample grouping, controls, and validation planning.
Step 7: Evaluate QC Readouts Before Scaling Up
Before scaling to a full cohort, evaluate a pilot PhIP-Seq run with multiple QC readouts: library diversity, recovered DNA yield, sequencing depth, read distribution, enrichment over negative controls, replicate correlation, and positive-control behavior.
Poor replicate overlap may indicate unstable binding, insufficient library representation, inconsistent bead capture, or too few reads. High background suggests nonspecific pull-down, inadequate blocking, or concentrated serum input. Low reads across all samples may point to library preparation, amplification, or sequencing issues.
A decision tree can keep troubleshooting systematic. Identify where signal is lost: before binding, during capture, during washing, or during sequencing library preparation. Changing all conditions at once may produce a usable result, but it rarely explains why the first experiment failed.
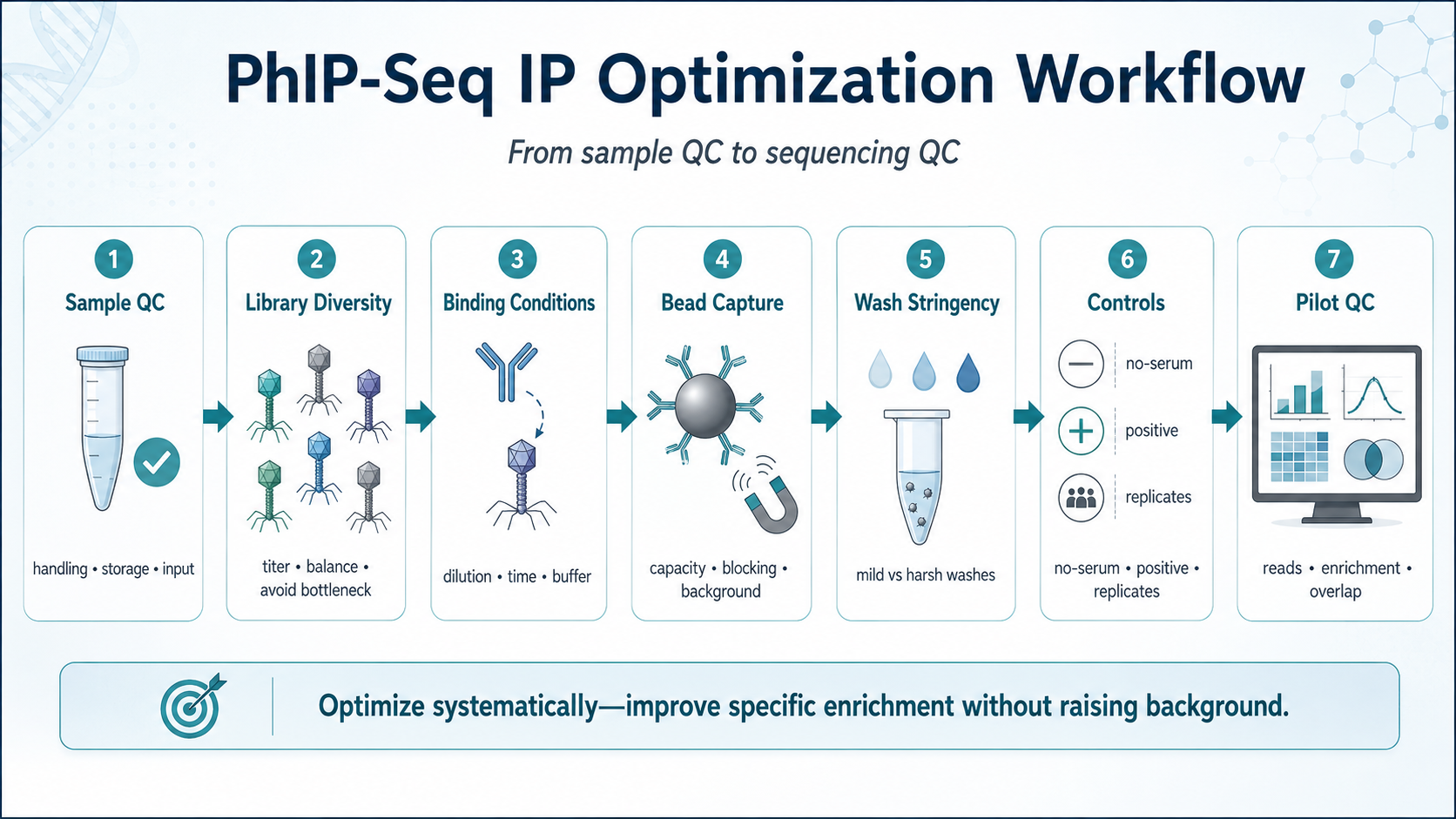
Figure 2. PhIP-Seq IP optimization workflow from sample QC to sequencing QC.
Common Mistakes to Avoid
One common mistake is treating IP performance as a yield-only problem. More recovered material does not always mean better data if sequencing depth is spent on nonspecific background.
Another mistake is changing too many parameters in one round. Changing serum dilution, bead amount, wash detergent, and incubation time together may improve the result, but it obscures which factor mattered.
Researchers should also avoid over-interpreting single-sample hits. Candidate peptides should be evaluated against controls, replicates, and biological metadata. Strong candidates still require validation with targeted methods such as ELISA, peptide arrays, or Western blotting.
FAQ
1. What is a good indicator of improved PhIP-Seq immunoprecipitation efficiency?
Improved efficiency should show stronger enrichment over negative controls, acceptable recovered material, stable replicate overlap, and preserved library diversity. DNA yield alone is not enough.
2. Should serum dilution be optimized for every PhIP-Seq project?
Yes, especially when sample type, disease background, antibody abundance, or expected signal strength is uncertain. A small dilution series can help balance specific signal and nonspecific background.
3. Can stricter washing improve PhIP-Seq data quality?
Stricter washing can reduce background, but it may also remove weak binders. Evaluate wash conditions with positive controls, negative controls, and replicate consistency.
4. Why do PhIP-Seq replicates sometimes show poor overlap?
Poor overlap may result from low library representation, unstable binding, inconsistent bead capture, insufficient sequencing depth, or sample handling variation. Diagnose this before scaling the assay.
Conclusion
Optimizing PhIP-Seq immunoprecipitation efficiency requires a systematic view of the workflow. Sample quality, library diversity, antibody binding, bead capture, wash stringency, controls, and QC readouts all influence the final enrichment profile.
For teams using PhIP-Seq in serum antibody profiling, epitope discovery, vaccine response, or biomarker screening, workflow review can prevent sample loss and repeat experiments. To plan a pilot study or troubleshoot weak enrichment data, contact MtoZ Biolabs to evaluate sample readiness, capture conditions, controls, and validation options before the full cohort is processed.
How to order?

