





How to Optimize DDA Parameters for High-Quality Protein Identification?
-
Maximizing sampling of peptide diversity to improve proteome coverage.
-
Improving detectability of low-abundance proteins.
-
Reducing redundant MS2 events and background interference to improve spectrum quality.
-
Enhancing reproducibility across experiments.
-
Minimize protein degradation: add protease inhibitors and maintain low temperatures throughout processing.
-
Optimize proteolysis conditions: including trypsin-to-protein ratio, digestion temperature, and incubation time.
-
Purify peptides using C18 cartridges to remove salts and other interfering components.
-
Quantify loading: ensure the same protein amount is injected in each run; BCA-based quantification is recommended.
-
For highly complex samples: increase N appropriately (e.g., Top 20).
-
To reduce repeated selection of the same precursors, enable dynamic exclusion.
-
Fixed NCE: 27-30%.
-
Stepped/variable NCE (recommended): automatically adapts to precursor m/z to improve fragment ion coverage.
-
Gradient duration: 60-120 min (longer gradients generally improve separation for complex samples).
-
Column selection: nano-flow C18 columns with 75 μm inner diameter and 1.9-3 μm particle size.
-
Flow rate: typically maintained below 300 nL/min.
-
Highly reproducible qualitative proteome profiling workflows.
-
Enrichment strategies and accurate identification for low-abundance proteins.
-
A fully traceable quality control framework spanning the entire workflow.
Data-dependent acquisition (DDA) remains one of the most widely used mass spectrometry acquisition strategies in proteomics. Although data-independent acquisition (DIA) has advanced rapidly in recent years, DDA continues to be essential for exploratory studies, spectral library and database construction, and high-resolution identification-focused proteomic analyses. Achieving high-quality protein identification in DDA experiments critically depends on systematic optimization of acquisition parameters.
Why Is DDA Parameter Optimization So Critical?
The core principle of DDA is to select the top N precursor ions by intensity from each MS1 survey scan for subsequent MS2 fragmentation. This strategy offers high specificity and efficient database searching, but it is also susceptible to sampling bias (e.g., preferential selection of high-abundance peptides).
Optimizing DDA parameters is important for:
Sample Preparation: The Basis of High-Quality Data
Prior to any instrumental optimization, sample quality is the primary determinant of DDA performance. Even with state-of-the-art instrumentation and well-chosen parameters, issues such as protein degradation, incomplete digestion, or inconsistent loading can substantially compromise identification outcomes.
Recommended practices:
MS1 Parameter Optimization: Ensure Accurate Precursor Ion Measurement
1. Resolution
(1) Recommended setting: 60,000-120,000 (Orbitrap platforms as an example).
(2) Optimization goal: improve peak capacity and precursor separation, which is particularly beneficial for resolving closely eluting peptides in complex samples.
2. Scan Range (m/z)
(1) Common range: 350-1600 m/z.
(2) Optimization strategy: adjust based on peptide size distribution; for samples enriched in shorter peptides, the lower bound can be reduced to 300 m/z.
3. Automatic Gain Control (AGC Target) and Maximum Injection Time (Max IT)
(1) AGC Target: determines the ion population accumulated prior to detection.
(2) Max IT: sets the upper limit for ion accumulation time.
(3) Recommended optimization: AGC 1e6 with Max IT 50-100 ms can improve spectral signal-to-noise while limiting background contributions.
MS2 Parameter Optimization: Improve the Informativeness of Fragment Spectra
1. Top N (TopN / Top Speed Mode)
(1) Definition: select the top N most intense precursor ions following each MS1 scan for MS2 fragmentation.
(2) Typical settings: Top 10-20.
(3) Optimization recommendations:
2. Dynamic Exclusion
(1) Purpose: prevent repeated fragmentation of the same precursor within a short time window.
(2) Recommended setting: 30-60 s.
(3) Expected impact: increases opportunities to sample lower-abundance precursors and can increase the number of identified proteins.
3. Fragmentation Mode and Collision Energy
(1) Common mode: HCD (higher-energy collisional dissociation).
(2) NCE optimization:
4. MS2 Resolution and Injection Time
(1) Recommended resolution: 15,000-30,000 to balance scan speed and spectral quality.
(2) Recommended Max IT: 50-100 ms to avoid excessively long accumulation times that reduce duty cycle.
LC Gradient Settings: Chromatographic Separation as the Front Line of MS
Even with well-optimized MS parameters, inadequate chromatographic separation of complex peptide mixtures can become a primary bottleneck for identification depth.
Recommendations for LC optimization:
In proteomics analyses, MtoZ Biolabs applies nano-flow gradient optimization strategies. By combining extended gradients with high-performance, high-resolution column configurations, this approach supports improved separation and enhanced detection of low-abundance peptides and proteins.
How to Evaluate the Effect of DDA Parameter Optimization?
Whether optimized settings are effective should be assessed using objective, quantitative readouts:
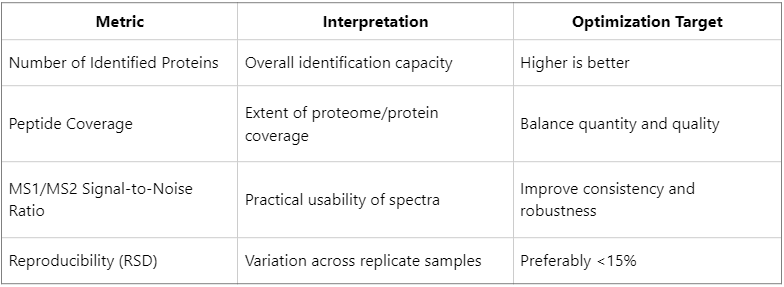
MtoZ Biolabs' DDA Optimization Solution
In practice, DDA parameter optimization often requires iterative tuning across diverse sample types (e.g., cells, tissues, plasma, and exosomes) and study objectives (e.g., differential expression, interactomics, and post-translational modification analyses). Drawing on experience from more than 10,000 DDA projects, MtoZ Biolabs has established a customized DDA parameter library organized by sample type + instrument platform + objective.
We utilize high-resolution mass spectrometry platforms, including Orbitrap Exploris 480 and Q Exactive HF-X, together with internally developed digestion and cleanup workflows, LC separation gradients, and analytical methods, to provide:
DDA parameter optimization is not an isolated adjustment, but a system-level workflow spanning sample preparation, LC-MS configuration, and acquisition strategy design. Through evidence-based parameter tuning, researchers can improve both the depth and reliability of protein identification while strengthening experimental stability and reproducibility. For studies requiring high-quality, deep-coverage, and highly reproducible proteomics data, you are welcome to contact MtoZ Biolabs; our technical team and platform capabilities are positioned to support your research objectives.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

