





How to Identify Protein–Protein Interactions Using Mass Spectrometry?
- Fuse the protein of interest to a tagged expression construct (e.g., Flag, HA, Strep-tag, etc.)
- Lyse cells and perform immunoaffinity enrichment using the corresponding antibody
- Elute the captured complexes and subject them to MS to identify co-purified proteins
- High specificity and experimentally straightforward implementation
- Compatible with stable cell lines or transient expression systems
- Can be combined with tag-based quantitative methods to compare interaction strength across conditions
- May fail to capture transient or weak-affinity interactions
- Tag placement can potentially affect protein structure and function
- Provides residue-level interaction-site information
- Enables conformational analysis of protein complexes
- Applicable to in situ cellular contexts and tissue samples
- Complex downstream data analysis and interpretation
- Requires high-resolution MS and strong algorithmic support for data processing
- Well suited for weak or transient interactions
- Supports detection in living cells under in situ conditions
- Less dependent on the stability of purified structural complexes
- Relatively high background labeling can occur
- Labeling duration and expression levels typically require optimization
- Design and construction of expression vectors
- Cell-line generation and validation of tag expression
- Purification and enrichment options across multiple strategies
- High-resolution MS analysis using Orbitrap Exploris and QE Plus platforms
- End-to-end workflows for quantitative, crosslinking, and proximity-labeling interaction analyses
- Bioinformatics support for spectral interpretation and interaction-network construction
Protein–protein interactions (PPIs) are central to biological processes such as cellular signal transduction, metabolic regulation, and chromatin remodeling. Identifying and characterizing protein interaction networks not only facilitates mechanistic understanding of cellular functions but also plays an important role in drug-target screening and studies of disease mechanisms. With the rapid advancement of mass spectrometry (MS) in proteomics, MS has become one of the most widely used and robust approaches for investigating PPIs.
How Does Mass Spectrometry Enable Studies of Protein–Protein Interactions?
Although traditional yeast two-hybrid (Y2H) and co-immunoprecipitation (Co-IP) assays are useful for detecting PPIs, each has clear limitations: Y2H is often biased toward interactions that can be reconstituted in the yeast nuclear environment, whereas Co-IP can suffer from non-specific background signals. By contrast, MS integrated with immunoaffinity enrichment, chemical crosslinking, and related strategies can support high-throughput, quantitative, and structure-informative analyses of protein interactions.
Key advantages include:
(1) High-throughput identification: a single experiment can identify hundreds to thousands of interacting proteins.
(2) High specificity: tag-based purification, stringent washing/elution procedures, and appropriate controls can substantially reduce false positives.
(3) Strong quantitative capability: coupling with labeling strategies such as SILAC and TMT enables dynamic assessment of interaction strength.
(4) Structural insight: crosslinking MS can provide information on interaction sites and spatial conformational constraints.
Mainstream MS-Based Strategies for Protein–Protein Interactions Analysis
1. Affinity Purification Mass Spectrometry (Affinity Purification–MS, AP-MS)
AP-MS is currently among the most commonly used approaches for interaction studies. The general workflow includes:
(1) Advantages
(2) Limitations
MtoZ Biolabs employs a low-background magnetic bead system together with an optimized lysis/elution workflow, and leverages an Orbitrap high-resolution MS platform to enable accurate identification and quantification of low-abundance interacting proteins.
2. Cross-Linking Mass Spectrometry (Cross-Linking MS, XL-MS)
In XL-MS, chemical crosslinkers are used to “lock” spatially proximal amino acid residues. Following proteolytic digestion, enrichment, and MS analysis, XL-MS can identify interacting proteins while also providing interaction-site information at the structural level.
(1) Advantages
(2) Technical challenges
MtoZ Biolabs integrates commonly used crosslinkers such as DSS and BS3, together with a dedicated workflow for enriching crosslinked peptides and in-house analysis algorithms, thereby improving crosslinked-spectrum identification rates and the accuracy of site-level interpretation.
3. Tag-Assisted Proximity Labeling (BioID / TurboID)
BioID and TurboID exploit proximity-labeling by fusion proteins. A mutant biotin ligase labels proximal proteins with biotin in living cells, after which streptavidin-based affinity purification followed by MS is used to identify interacting proteins.
(1) Advantages
(2) Notes
MtoZ Biolabs has established a TurboID lentiviral platform for generating stable cell lines, supported by biotin enrichment and deep MS detection workflows, which is suitable for hard-to-transfect cells or primary cell lines.
Recommendations for Choosing Among Strategies
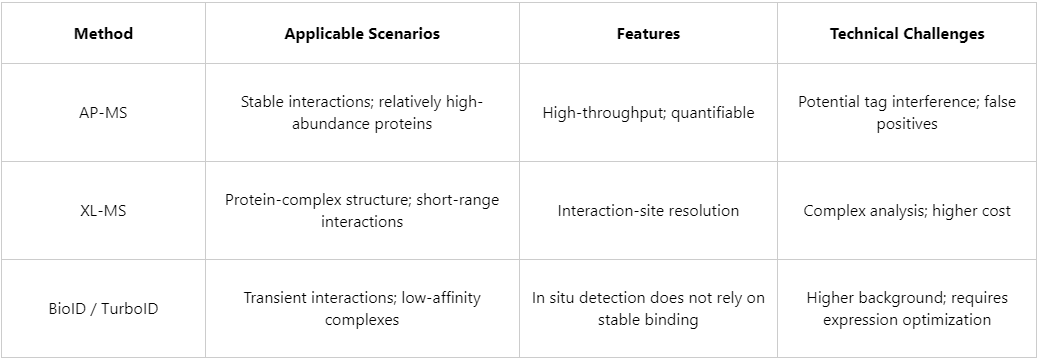
Depending on the research objective, combining multiple strategies is often beneficial. For example, TurboID can be used to screen candidate interactors, AP-MS can then be applied to validate specificity, and XL-MS can be used to further resolve structural features.
Advantages of MtoZ Biolabs' Mass Spectrometry Services for Protein–Protein Interactions Studies
As a specialized proteomics and MS platform, MtoZ Biolabs provides an integrated solution for PPI studies, including:
Whether the goal is discovery of novel drug targets or mechanistic dissection of complex signaling pathways, we can provide customized, high-sensitivity, publication-ready MS datasets to support protein-interaction studies.
Mass Spectrometry has evolved from a tool primarily used for protein identification into a core methodology for interrogating dynamic interaction networks and structural conformations. As AP-MS, XL-MS, and TurboID continue to mature, researchers can more comprehensively elucidate the molecular cooperation underlying cellular function. If you are planning protein–protein interactions studies, you are welcome to contact MtoZ Biolabs for a complimentary technical consultation and project evaluation. We look forward to advancing life-science research together, step by step.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

