





How to Detect Protein Acetylation Using Mass Spectrometry?
-
Diverse modification sites: widely distributed across transcription factors, histones, metabolic enzymes, and other proteins.
-
Low abundance and high dynamics: Kac modifications are present at extremely low abundance within the total proteome (<0.1%).
-
Defined mass shift: +42.0106 Da, which can be accurately detected by high-resolution mass spectrometry.
-
High positional specificity requirement: precise localization to specific lysine residues is essential.
-
Lysis buffers containing deacetylase inhibitors (e.g., Trichostatin A and Nicotinamide) are used to preserve endogenous acetylation modifications.
-
An 8 M urea or SDS-based lysis buffer system is recommended for efficient protein extraction.
-
Trypsin is routinely employed for proteolytic digestion to generate peptides suitable for subsequent enrichment and analysis.
-
Antibodies specifically recognize the covalent structure of acetylated lysine residues.
-
Applicable to diverse sample types, including cells, tissues, and clinical specimens.
-
Can be integrated with subcellular fractionation strategies or combined with isobaric labeling methods (e.g., TMT).
-
High-resolution Orbitrap series instruments (e.g., QE HF-X, Exploris 480)
-
timsTOF Pro 2 is suitable for DIA acquisition modes.
-
DDA (data-dependent acquisition): suitable for comprehensive site mapping
-
DIA (data-independent acquisition): suitable for quantitative analysis of large sample cohorts
-
Set acetylation (K+42.0106) as a variable modification during database search.
-
MS/MS fragmentation scans are required to confirm modification site localization.
-
Dynamic exclusion should be enabled to avoid repeated fragmentation of highly abundant peptides.
-
Label-based quantification: TMT, iTRAQ, etc
-
Label-free quantification: LFQ or MS1 peak area-based quantification
Protein acetylation is one of the earliest discovered and most extensively studied forms of acylation, particularly lysine acetylation (Kac). It is widely present in nuclear, cytoplasmic, and mitochondrial proteins and plays critical roles in transcriptional regulation, chromatin remodeling, and metabolic control. With the advancement of mass spectrometry (MS) technologies, researchers can now perform high-throughput, quantitative, and site-specific analyses of acetylation at the proteome-wide level.
Characteristics and Analytical Challenges of Lysine Acetylation
Therefore, detection strategies with high sensitivity and specificity are critical for successful investigation of Kac.
Standard Workflow for Mass Spectrometry-Based Detection of Acetylation
1. Protein Extraction and Digestion
2. Enrichment of Acetylated Peptides (Key Step)
Given the low stoichiometry of acetylation, enrichment prior to MS analysis is essential:
Common approach: anti-acetyl-lysine antibody-based immunoprecipitation (Anti-Kac IP).
3. LC-MS/MS Analysis
(1) Liquid chromatography separation: NanoLC systems improve separation efficiency; C18 reversed-phase columns are recommended.
(2) Recommended mass spectrometry platforms
(3) Acquisition strategies
(4) Instrument parameter settings
4. Data Analysis and Site Localization

Data analysis workflow
(1) Database search: use the UniProt species-specific database, setting Kac as a variable modification.
(2) Site localization: apply ptmRS or Ascore algorithms to evaluate site confidence (typically requiring localization probability ≥ 0.75).
(3) Quantitative analysis
Advanced Strategies to Enhance Depth and Throughput in Acetylation Proteomics
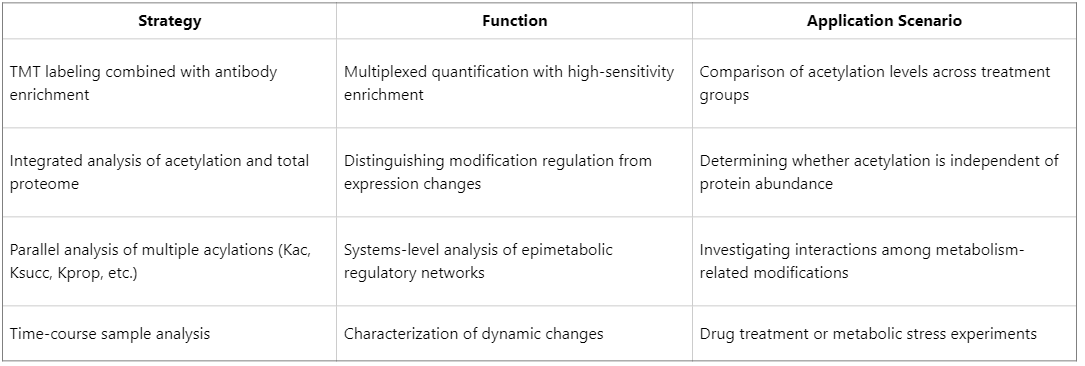
Major Application Areas of Protein Acetylation Research

How to order?

