





DIA vs DDA: A Comparative Overview of Data Acquisition Principles in Phosphoproteomics
-
Generates high-quality MS/MS spectra, which are well suited for confident peptide identification and spectral library construction.
-
Supported by a mature software ecosystem, enabling reliable database searching and de novo peptide sequencing.
-
Strongly biased toward high-abundance peptides, leading to frequent omission of low-abundance phosphopeptides.
-
Limited reproducibility, as the selected MS2 precursors may vary between runs or batches.
-
High stochasticity, which restricts its applicability in large-scale quantitative comparisons.
-
Provides comprehensive and consistent sampling of peptides, with enhanced sensitivity for low-abundance phosphopeptides.
-
Exhibits superior reproducibility and quantitative accuracy, making it well suited for large-cohort and high-throughput studies.
-
Enables retrospective data re-analysis, thereby offering strong data reusability and long-term analytical value.
-
Data interpretation is computationally intensive and typically relies on high-quality spectral libraries or advanced AI-based algorithms.
-
Method development and data processing require greater computational resources and technical expertise.
-
DDA is more appropriate for samples with relatively simple backgrounds and abundant phosphopeptides.
-
DIA enables more comprehensive identification of low-abundance phosphopeptides in complex biological samples and demonstrates superior performance in signaling pathway-focused studies.
-
DDA often exhibits substantial variability across experimental batches.
-
DIA provides highly consistent quantitative results and is particularly suitable for multi-omics integration, time-course analyses, and large-scale cohort studies.
-
DDA captures only a subset of fragment ion spectra, limiting the feasibility of downstream re-analysis.
-
DIA records comprehensive fragment ion information across the entire mass range, allowing data re-interrogation with newly developed analytical algorithms and extending the overall data lifecycle.
-
Construction of high-quality phosphoproteomic spectral libraries using DDA.
-
Large-scale and high-throughput phosphoproteomic quantification enabled by DIA acquisition.
-
Implementation of advanced AI-driven tools (e.g., DIA-NN and Spectronaut) to support library-free DIA data analysis.
-
End-to-end services encompassing phosphopeptide enrichment, quantitative analysis, and signaling pathway interpretation.
Protein phosphorylation is one of the most prevalent and functionally important post-translational modifications (PTMs) involved in cellular signal transduction and regulatory processes. In proteomics research, mass spectrometry (MS) serves as the central analytical tool for the identification of phosphorylation sites. Among available MS data acquisition strategies, data-dependent acquisition (DDA) and data-independent acquisition (DIA) are the two most widely used approaches. These strategies differ substantially in phosphopeptide identification depth, quantitative accuracy, and analytical reproducibility.
Comparison of Technical Principles: DDA vs DIA
1. DDA (Data-Dependent Acquisition)
DDA is a signal-intensity-driven acquisition strategy. Following a full MS1 survey scan, the mass spectrometer automatically selects the top N most intense precursor ions for fragmentation (MS2), generating tandem mass spectra for peptide identification.
(1) Advantages
(2) Disadvantages
2. DIA (Data-Independent Acquisition)
DIA adopts a systematic and unbiased acquisition strategy. The MS1 mass range is segmented into consecutive isolation windows, within which all precursor ions are simultaneously fragmented and recorded as MS2 spectra.
(1) Advantages
(2) Disadvantages
Application-Level Differences in Phosphoproteomics
1. Phosphopeptide Identification Performance
2. Reproducibility and Quantitative Accuracy
3. Data Reusability
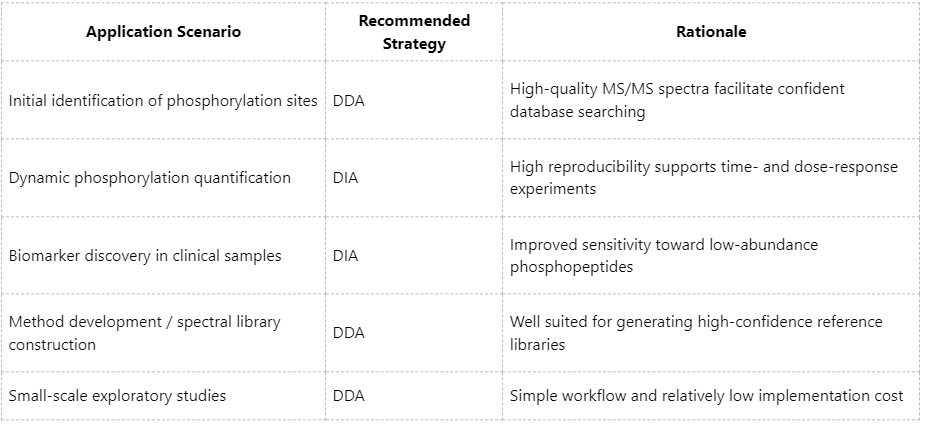
Practical Considerations for Method Selection
Phosphoproteomics Solutions at MtoZ Biolabs
In phosphoproteomics research, MtoZ Biolabs strategically integrates the complementary strengths of DDA and DIA to deliver flexible and robust analytical solutions:
The DIA platform at MtoZ Biolabs, built upon high-resolution Orbitrap mass spectrometers and independently optimized sample preparation workflows, enables the identification of more than 10,000 phosphorylation sites in a single experiment. This platform has been extensively applied to studies of signaling pathway regulation, drug target discovery, and clinical biomarker exploration.
In summary, both DDA and DIA offer distinct advantages in phosphoproteomics, and the optimal strategy should be selected based on specific research objectives, sample scale, budget considerations, and data analysis capabilities. For researchers seeking in-depth characterization of signaling networks, dynamic monitoring of kinase activity, or systematic phosphoproteomic biomarker discovery, DIA is increasingly recognized as a highly efficient, reproducible, and scalable solution. At MtoZ Biolabs, we provide comprehensive DDA- and DIA-based phosphoproteomics workflows to support the entire research pipeline, from phosphorylation site discovery to mechanistic interpretation. For further details regarding our service processes and representative case studies, we welcome inquiries to our technical team.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

