





Common PhIP-Seq Troubleshooting Questions: Weak Enrichment, Background Signals, and Library Coverage
- whether expected positive samples are truly underperforming
- whether reported peptide hits are inflated by background signal
- whether sequencing depth is being spent on a narrow peptide subset
- whether antigen-level aggregation remains interpretable
- whether the next step should be reanalysis, rerun, redesign, or orthogonal validation
- controls carrying many of the same top peptides as test samples
- poor signal-to-background ratio for known positive control peptides
- batch-specific shifts in control count distribution
- broad low-level enrichment across many peptides rather than a focused set
- percentage of library peptides detected above a minimum count threshold
- distribution of peptide-level counts across the library
- dominance of highly abundant peptides that compress dynamic range
- fraction of reads mapping to library members
- consistency of detected peptide breadth across replicates and batches
- whether top hits remain after alternate normalization choices
- whether replicate concordance improves after antigen-level aggregation
- whether enriched peptides cluster in biologically plausible antigen regions
- whether significance calls are driven mainly by a single replicate or batch
Weak enrichment, background-heavy controls, and patchy library coverage in a PhIP-Seq project should be treated first as a QC interpretation problem, not as proof that the biology is absent or that the dataset has no value. The most useful starting check is to compare four items side by side: input library / library reference representation, negative control behavior, sequencing depth distribution, and replicate concordance. That side-by-side view often shows whether the main constraint comes from assay setup, library performance, read allocation, or analysis thresholds.
In practice, teams should avoid repeating the full experiment until they know which layer is failing. Weak enrichment with good read mapping but a poor signal-to-background ratio often points to immunoprecipitation stringency or thresholding. Broad background signal in a mock IP / bead-only control often points to nonspecific pull-down or recurrent sticky peptides. Sparse peptide-level counts with uneven peptide representation more often indicate library dropout, compressed sequencing depth, or zero inflation that makes enrichment calls unstable.
Where These Problems Usually Appear
These issues usually show up after a pilot run, an initial cohort batch, or a vendor report review. The team expects reactivity to known antigens, yet enrichment above controls is modest. At the same time, negative control samples retain appreciable reads, some peptides dominate the count distribution, and other regions of the phage-displayed peptide library appear nearly absent.
The operational problem is not simply “low signal.” It affects several decisions at once:
If this is not resolved early, teams may validate false positives, miss real linear epitope-associated patterns, or repeat wet-lab work when the main bottleneck was elsewhere.
Root-Cause Map for Weak PhIP-Seq Performance
Most report-level failures in this scenario fall into four categories.
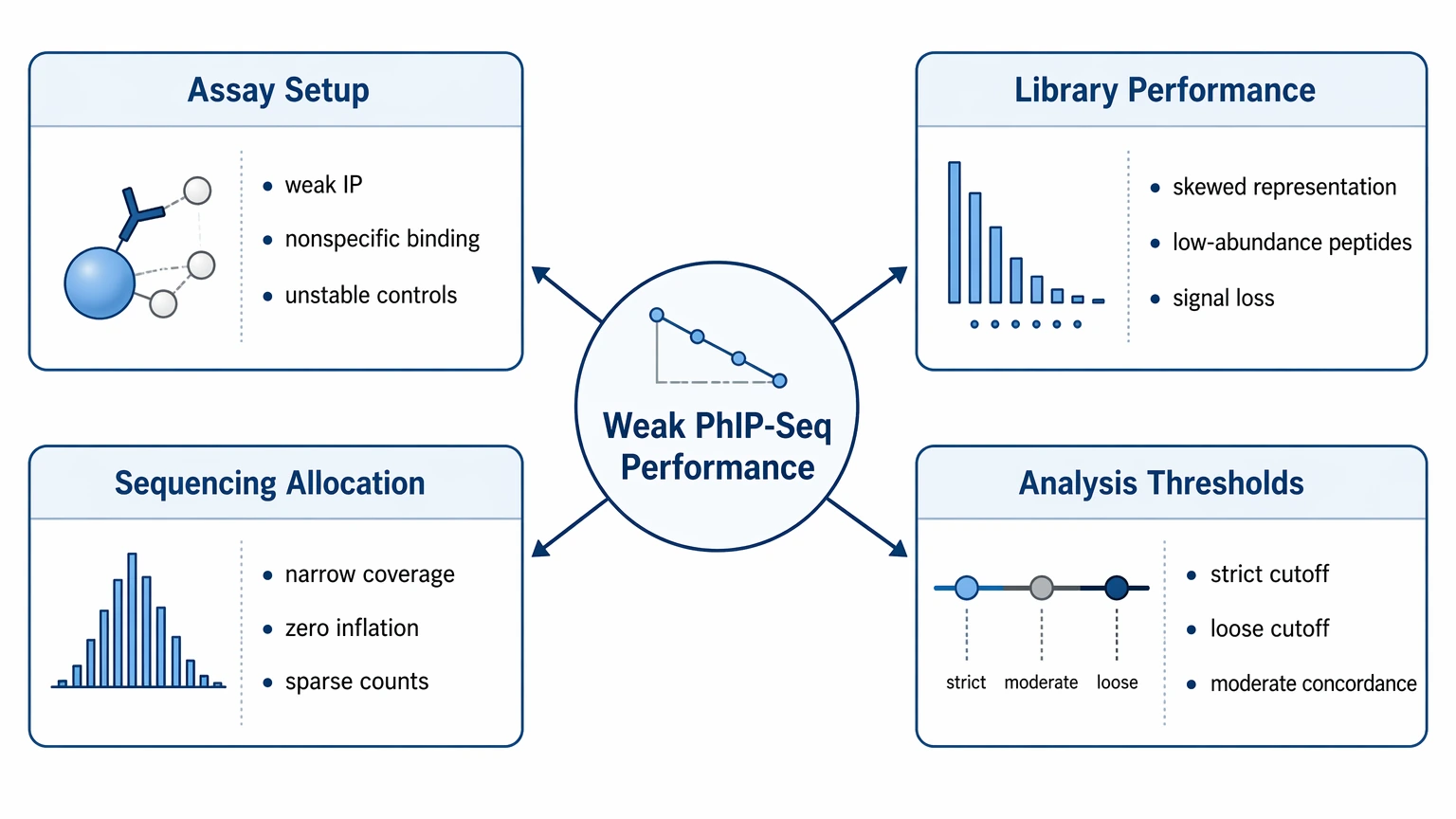
1. Assay setup problems flatten enrichment
Weak recovery during immunoprecipitation, excessive nonspecific binding, or unstable control structure can reduce enrichment and raise background signal. In reports, this usually appears as poor separation between study samples and the negative control.
2. Library performance creates hidden signal loss
If the input library / library reference already shows skewed peptide representation, low-abundance members may never receive enough reads to support stable enrichment statistics. That can look like biological absence even when antibodies are present.
3. Sequencing allocation narrows usable coverage
Adequate total reads do not guarantee usable library coverage. When mapped reads cluster in a small peptide subset, the rest of the library becomes sparse, zero inflation increases, and peptide-level counts get harder to interpret.
4. Analysis thresholds hide or inflate findings
Overly conservative normalization or enrichment cutoffs can suppress weak but reproducible binders. Loose thresholds can turn control-like noise into false positives. This becomes more likely when replicate concordance is only moderate.
A Step-by-Step Troubleshooting Path for PhIP-Seq Analysis
Step 1: Define what “weak enrichment” means in your report
Start by separating low read count, low fold-change, and low statistical confidence. These are different failure modes, and they should not be lumped together. Review total reads per sample, usable reads after preprocessing, fraction of reads retained after read mapping, and enrichment outputs such as fold-change, z-score, or model-based significance values.
A sample can have acceptable sequencing depth and still show weak enrichment if expected binders are diluted by background signal. Another sample may show strong-looking fold-change from a very low baseline yet remain unstable because peptide-level counts are sparse. End this step by naming the specific failure: low abundance, weak separation from controls, or low confidence after normalization.
Step 2: Read the controls before interpreting biology
Next, inspect the negative control structure before focusing on apparent hits. A mock IP / bead-only control should show whether background signal is broad, recurrent, or driven by a specific set of sticky peptides. If the same peptides repeatedly enrich in controls across batches, changing cutoffs alone rarely fixes the problem.
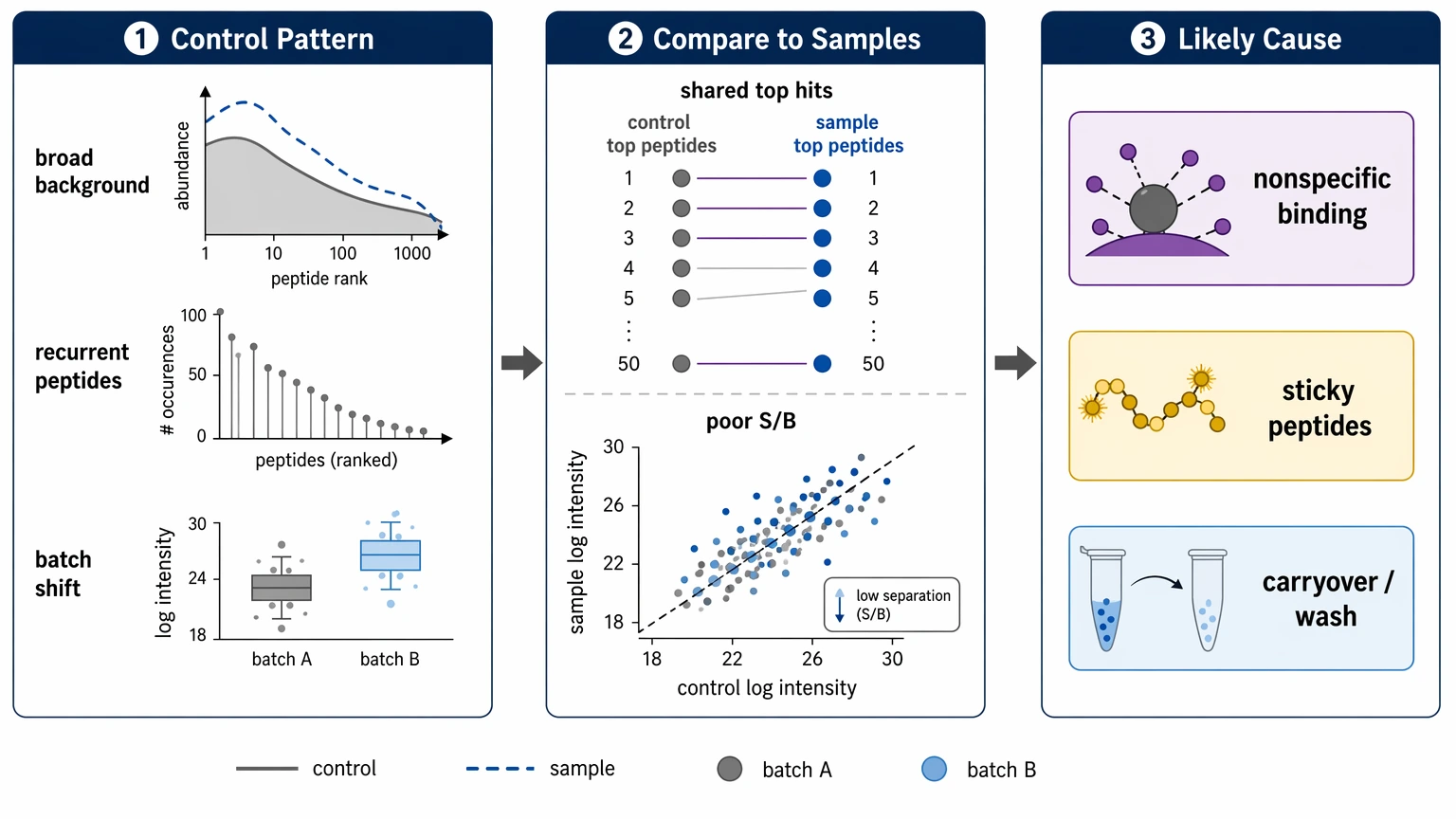
Look for these patterns:
These patterns point to nonspecific binding, bead/phage carryover, or insufficient wash stringency. If your report does not show control distributions, recurrent control peptides, and sample-to-control contrast, ask for that review before trusting peptide-level conclusions. If the next decision is whether to rerun or reinterpret the dataset, you can submit your requirements to MtoZ Biolabs for a PhIP-Seq project evaluation focused on control behavior, library reference balance, and enrichment outputs.
Step 3: Distinguish library dropout from shallow sequencing
Do not assume poor coverage can be fixed by adding more reads. First compare the input library / library reference with post-immunoprecipitation samples. If the library reference itself shows uneven peptide representation, missing regions, or strong dominance by a subset of members, the project may be affected by library dropout or library construction bias.
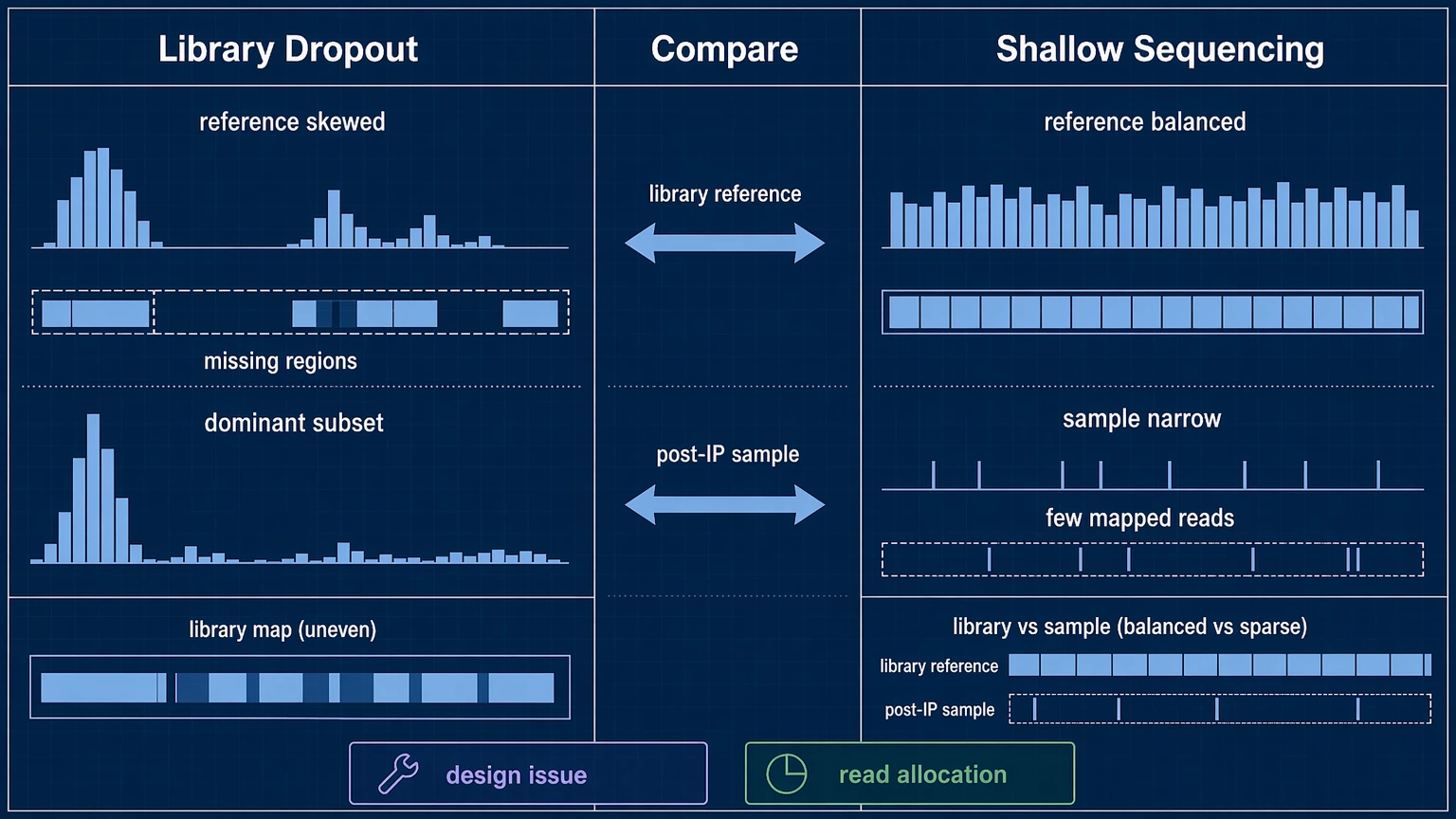
Then compare that baseline with sample-level library coverage:
If the library reference looks balanced but individual samples show narrow coverage, the main issue is more likely sequencing depth allocation or sample-specific pull-down behavior. If both the reference and the samples are skewed, repeating sequencing alone is unlikely to rescue interpretation.
Step 4: Check whether normalization is distorting the result
When controls are noisy and counts are sparse, normalization has a large effect on interpretation. Review whether the pipeline stabilizes low counts, handles zero inflation, and compares samples against the right negative control baseline. A rigid count cutoff can discard consistent weak binders, while a permissive threshold can turn stochastic reads into false positives.
Useful checks include:
This step should answer a practical question: does reanalysis materially change interpretation, or is the limitation upstream? If alternate models still fail to separate signal from background, the bottleneck is usually assay behavior, library performance, or read allocation rather than statistics alone.
Step 5: Choose the correction path before repeating the study
After the first four steps, choose one primary action.
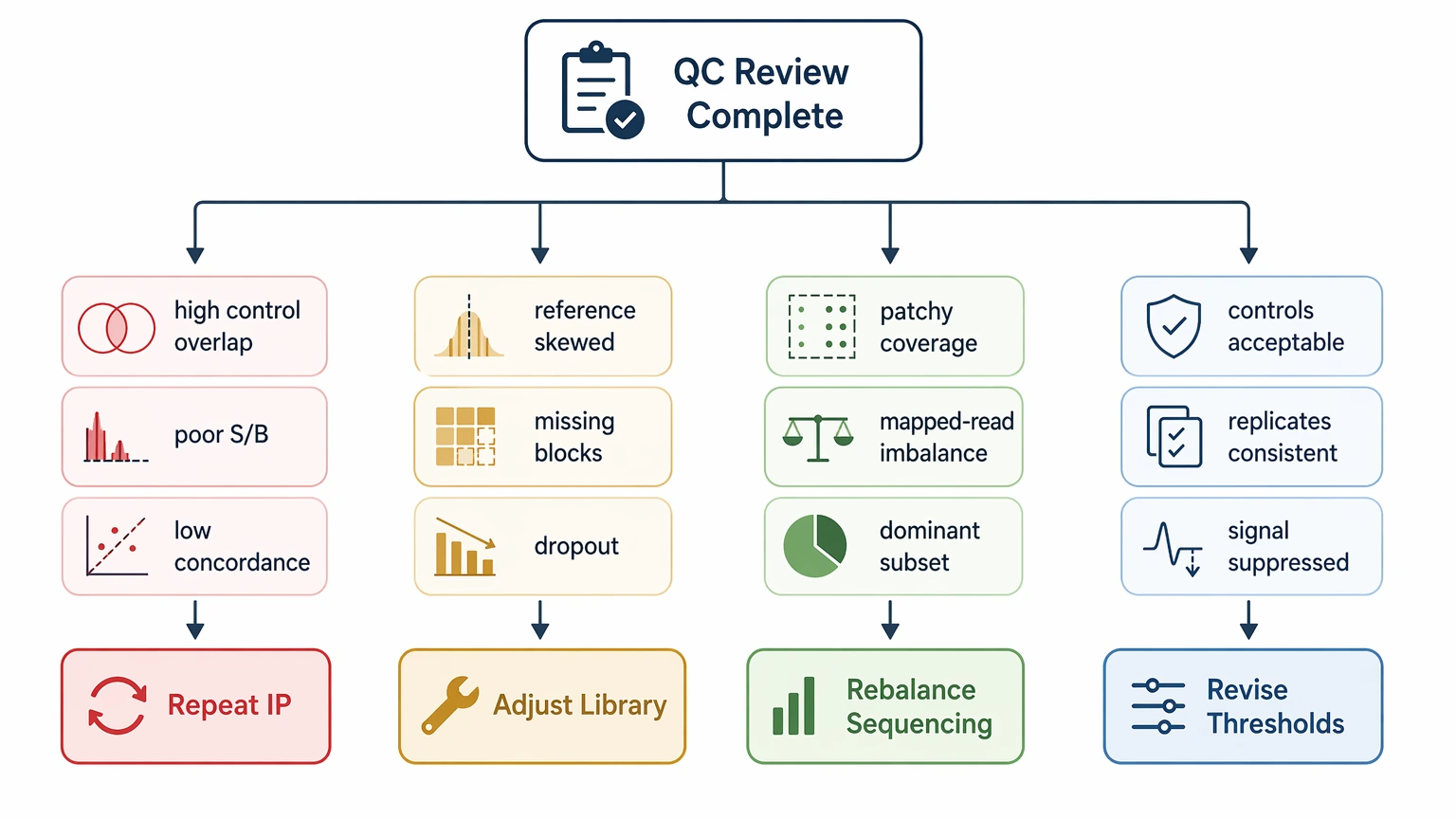
Repeat immunoprecipitation when controls show strong nonspecific signal, signal-to-background ratio is poor, or replicate concordance remains low despite reasonable library representation.
Adjust library design or sourcing when the input library / library reference already shows uneven peptide representation, missing peptide blocks, or clear evidence of library dropout.
Rebalance sequencing allocation when total project output looks adequate but sample-level library coverage is patchy, mapped reads vary sharply across samples, or a small peptide subset dominates the count distribution.
Revise analysis thresholds when controls are acceptable, replicates are reasonably consistent, and expected biology is visible but suppressed by conservative filtering.
If the dataset still contains a small set of repeatable, biologically coherent hits, it may support targeted follow-up rather than full discard. In that case, move the strongest candidates into orthogonal validation instead of treating every enriched peptide as equally credible.
Which QC Signals Should Trigger Rerun, Reanalysis, or Validation
Use project decisions, not single metrics, to judge readiness.
Rerun is more justified when the negative control overlaps heavily with study samples, the signal-to-background ratio remains weak for expected positives, or replicate concordance is poor at both peptide and antigen levels.
Reanalysis is more justified when mapped read fraction is acceptable, library coverage is reasonably broad, controls are interpretable, and the main uncertainty comes from normalization, cutoff choice, or low-count modeling.
Validation is more justified when a limited set of hits persists after control-aware filtering, appears across replicates, and supports a plausible antigen-level aggregation pattern. Those findings are better candidates for ELISA, peptide array testing, or other orthogonal validation than isolated single-peptide spikes.
Interpretation Limits That Matter in Practice
PhIP-Seq measures antibody binding to a phage-displayed peptide library, so it is better suited to linear epitope-associated interpretation than to broad claims about conformational binding. A peptide hit is not automatically equivalent to a confirmed antigen-level result, and a weak dataset does not prove antibody absence.
Batch effects matter too. If control distributions shift across runs or library lots behave differently, cohort-level patterns may reflect workflow structure rather than biology. For service buyers, a credible troubleshooting report should explain control behavior, sequencing allocation, normalization logic, and the limits of peptide-level conclusions.
Conclusion
Weak enrichment, elevated background signal, and uneven library coverage in PhIP-Seq become easier to interpret when assay behavior, phage-displayed peptide library performance, sequencing depth allocation, and normalization choices are reviewed separately rather than treated as one combined failure. The most reliable workflow is to review control structure, input library / library reference balance, count distribution, and replicate concordance before deciding to rerun, reanalyze, or validate selected hits.
This framework is especially useful for pilot studies, batch-scale antibody profiling projects, vendor report reviews, and datasets where peptide-level conclusions remain uncertain but not meaningless. If you need a technical review of control behavior, library coverage, or a follow-up orthogonal validation plan, contact MtoZ Biolabs to evaluate your project and discuss the most defensible next step before committing more samples or sequencing budget.
FAQ
What is the difference between weak enrichment and low sequencing depth in PhIP-Seq?
Low sequencing depth reduces the number of usable observations across the library. Weak enrichment means expected binders fail to separate clearly from the negative control even when read mapping and sample-level depth are acceptable. You need both coverage metrics and control contrast to tell them apart.
What control pattern most strongly suggests nonspecific binding?
A strong warning sign is repeated enrichment of the same peptides in mock IP / bead-only control samples across batches, especially when those peptides also appear among top hits in study samples. That pattern is more consistent with background signal than with sample-specific antibody reactivity.
How can I tell whether poor library coverage starts in the library or after immunoprecipitation?
Compare the input library / library reference with post-IP samples. If underrepresented regions are already missing in the reference, the issue likely starts with peptide representation or library dropout. If the reference is balanced but only certain samples show narrow coverage, sequencing allocation or pull-down behavior is the more likely cause.
When is antigen-level aggregation helpful, and when does it hide a problem?
Antigen-level aggregation is helpful when several related peptides move together across replicates and support a coherent pattern. It is less helpful when a reported antigen signal depends on one unstable peptide or one batch-specific outlier, because aggregation can mask weak underlying evidence.
What should I expect from a technically credible troubleshooting report?
It should show mapped read fraction, library coverage, control distributions, replicate concordance, normalization logic, and the rationale for any enrichment threshold. It should also state which findings are suitable for orthogonal validation and which remain too background-sensitive for confident interpretation.
How to order?

