





Stability of Proteins From Rates of Oxidation Analysis Service
To explore the local conformational function of proteins in relation to their native structural stability, Fitzgerald et al. developed a technique that utilized amide H/D exchange rates to detect protein amide groups. In this technique, the thermodynamic stability of proteins and protein ligand complexes was assessed by measuring their amide H/D exchange rate. This approach was later refined and evolved into stability of proteins from rates of oxidation (SPROX). In SPROX technology, hydrogen peroxide is added to oxidize proteins in the face of increasing concentrations of chemical denaturants. Each protein sample is reacted with the same amount of hydrogen peroxide for the same time in a buffer containing GdmCl. Traditional bottom-up proteome samples are then pretreated and mass spectrometry (MS) is used to determine the relative strength of unoxidized and oxidized methionine-containing peptides. The unoxidized and oxidized peptides containing arginine are quantified with the concentration of the SPROX buffered denaturant, resulting in the calculation of the folding free energy (ΔGf), binding free energy (ΔΔGf), and dissociation constant (KD) of the protein folding/unfolding reaction.
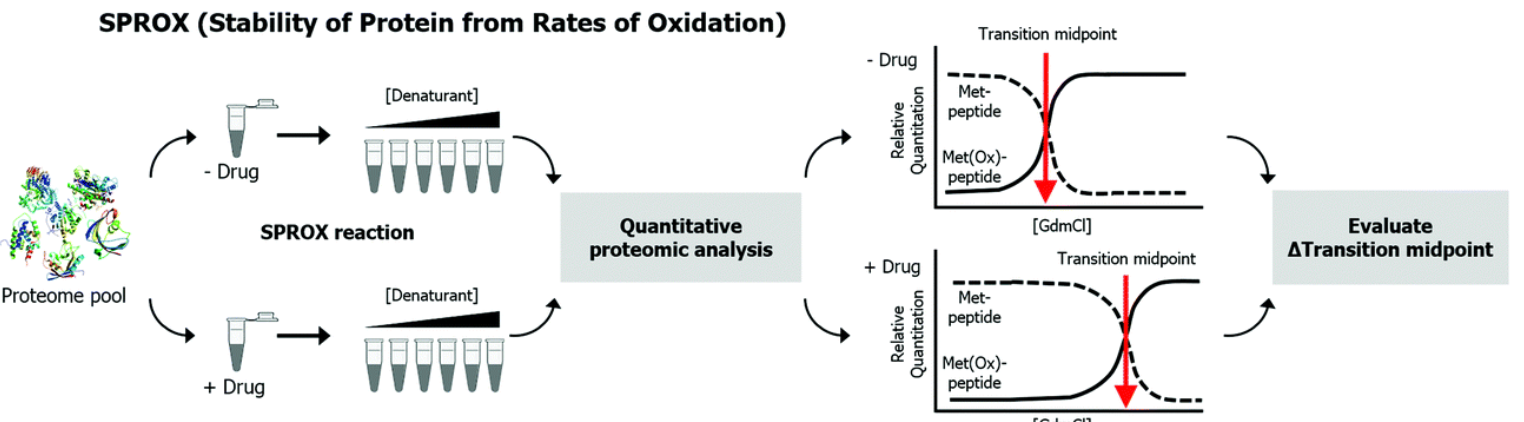
Figure 1. SPROX Technical Process [1]
The SPROX workflow is to divide complex biological mixtures of proteins (e.g., cell lysates) into two groups, one in the absence of drug and one in the presence of drug. For each analysis, an aliquot of the protein mixture (or the mixture of proteins binding to the drug) is diluted into a series of buffers containing increasing concentrations of chemical denaturants (e.g., guanidine hydrochloride (GdmCl)). The protein sample in each GdmCl-containing buffer reacts with the same amount of hydrogen peroxide for the same time. The reaction time and concentration of hydrogen peroxide are adjusted so that the thioether groups in the side chain of methionine residues are selectively oxidized. After the quenching oxidation reaction and the generation of trypsin peptides, tandem LC-MS/MS techniques, such as multidimensional protein identification technology (MudPIT), are applied to quantify the rate of selective methionine oxidation.
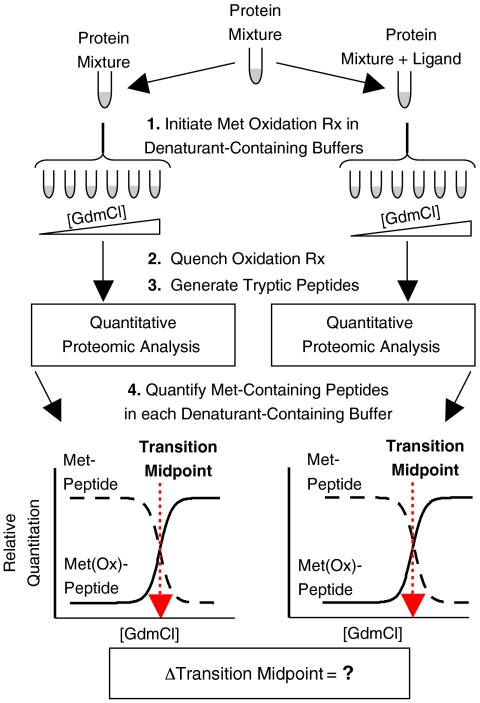
Figure 2. SPROX Technology Roadmap [2]
Although SPROX has similar advantages to MS-based thermal stability profiling and drug affinity reactive target stability (DARTS), one of the main limitations of SPROX is that it utilizes methionine oxidation as a parameter to measure thermodynamic properties, and all methionine residues do not necessarily exhibit different oxidation rates, providing sufficient information to confirm ligand-target interactions.
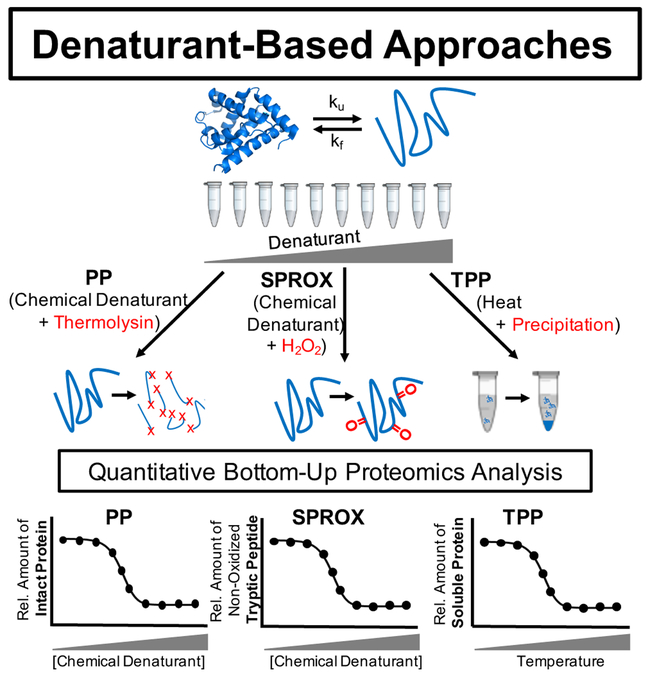
Figure 3. Comparison of SPROX Technology with Other Methods [3]
Combining SPROX with quantitative proteomic labeling technology expands the field of application of SPROX. SPROX is combined with stable isotope labeling with amino acids in cell culture (SILAC) to expand the coverage of SPROX on the whole proteome by labeling protein amino groups to determine the relative content of peptides. SPROX can also be combined with chemical labeling, in which protein samples in each buffer solution containing denaturant are precipitated in the solution after the oxidation reaction is quenched, and the precipitated protein samples are subjected to quantitative bottom-up proteomic analysis. In this strategy, precipitated protein samples from each denaturant-containing buffer are submitted to bottom-up proteomics sample preparation, including reduction of disulfide bonds, alkylation of cysteine side chains, trypsinization, and then labeling of peptides from different denaturant buffers with a set of isobaric mass tags (e.g, TMTsixplex™ or iTRAQ 8-Plex™). The isobaric mass labeling strategy reduces the number of LC-MS runs required in SPROX because aliquots from different denaturant-containing samples can be pooled and analyzed in a single run.
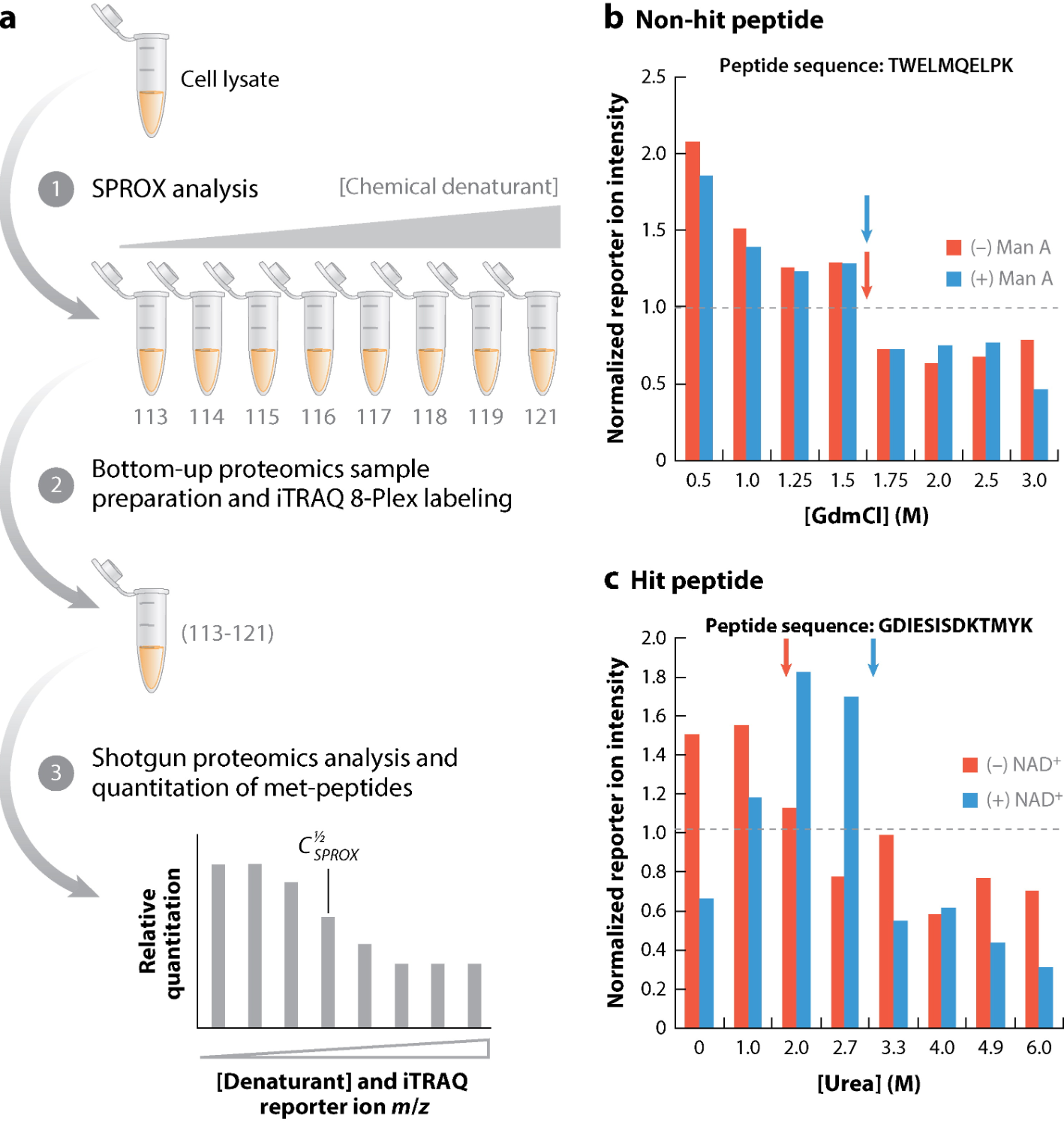
Figure 4. The Combination of SPROX and iTRAQ Technology [4]
Analysis Workflow
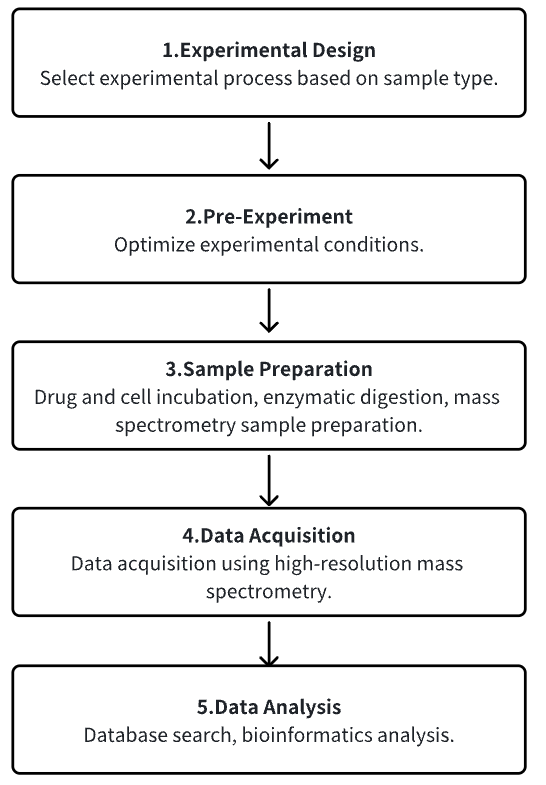
1. Determine the Experimental Process According to the Experimental Requirements
2. Pre-Experiment, Optimizing Experimental Conditions
3. Sample Preparation
4. High-Resolution MS Acquisition Data
5. Data Retrieval and Analysis
Service Advantages
1. Identification of Target Proteins from Multiple Types of Sample Sources, etc.
2. Provide Guidance on Optimizing Experimental Conditions
3. High-Confidence, High-Precision MS Detection
4. Comprehensive Bioinformatics Analysis
Sample Results
1. Analysis of Changes in Brain Protein Stability in Mouse Models of Normal Aging and α Synucleinopathy Reveals Age- and Disease-Related Differences
Parkinson's disease (PD) is a brain disorder that causes mobility problems as well as mental health, sleep, pain, and other health problems. SPROX was used to analyze the thermodynamic stability of proteins in brain tissue cell lysates from Huα-Syn (A53T) transgenic mice at three time points, including 1 month (n=9), 6 months (n=7), and onset of symptoms (between 9-16 months). Using ANOVA, the thermodynamic stability curves generated here on 332 proteins were compared with those generated on the same proteins from wild-type mice of similar age. This analysis identified a panel of 22 proteins with age-related changes in protein stability, and a panel of 11 proteins that were differentially stable in the Huα-Syn (A53T) transgenic mouse model. A total of 9 of the 11 proteins identified in the study had disease-related stability changes that had previously been detected in human cerebrospinal fluid and thus had potential utility as biomarkers of PD. The observed differential stability and age-related stability changes of a protein glutamate decarboxylase 2 (Gad2) were consistent with differences in known age-related truncates of the protein, which was shown here to have higher folding stability than full-length Gad2.
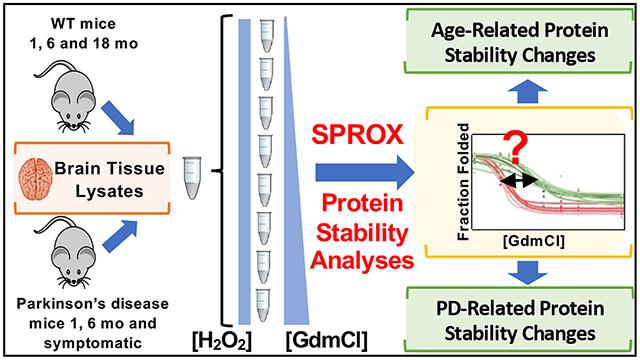
Figure 5. SPROX Reveals Changes in Brain Proteins Caused by Age and Disease [5]
2. Large-Scale Analysis of Conformational Changes in Breast Cancer-Associated Proteins Using SILAC-SPROX
Previous proteomic methods for disease state characterization and biomarker discovery utilize MS to identify proteins with altered expression levels in disease states. Large-scale use of protein folding stability measurements, stable isotope labeling with amino acids in cell culture and stability of proteins from rates of oxidation (SILAC-SPROX) has been reported to characterize different subtypes of breast cancer. Differences in protein folding stability were investigated in a comparison of luminal breast cancer subtypes, Luminal-A and -B (i.e., MCF-7 and BT-474 cells, respectively) and in a comparison of the luminal A and basal subtypes of disease (i.e., MCF-7 and MDA-MB-468 cells, respectively). In these comparative analyses, the stability of 242 and 445 protein hits changed, with a significant fraction of them showing no significant changes in expression levels. This suggested that thermodynamic stability measurements opened up a new avenue for protein biomarker discovery. From other biochemical studies, it is known that many identified protein hits played a role in tumorigenesis and cancer progression. This not only confirmed the biological significance of protein hits identified using the SILAC-SPROX, but also helped to elucidate the molecular basis of their dysregulation and/or dysfunction in cancer.
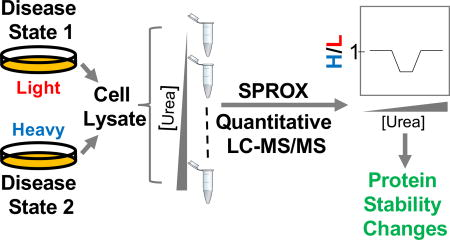
Figure 6. SILAC-SPROX Analysis of Protein Changes in Breast Cancer Cells [6]
3. The Malaria Parasite Chaperone Protein TRiC/CCT Was Identified as a Target for the Antihistamine Clomastine Using a Parallel Chemical Proteomics Strategy
There is an urging need for new drugs that target multiple stages of the malaria parasite to combat malaria resistance. The antihistamine clemastine inhibits multiple parasitic forms with unknown molecular targets. Parallel chemoproteomic platforms have been used to study the mechanism of action of clemastine based on MS-based thermal stability profiling and SPROX, and it has been found that clemastine binds to and destabilizes the plasmodium falciparum TCP-1 ring complex or chaperonin containing TCP-1 (TRiC/CCT), an essential heterooligomeric complexrequired for de novo cytoskeletal protein folding, which demonstrated that clemastine reduces levels of the major TRiC substrate tubulin in P. falciparum parasites.
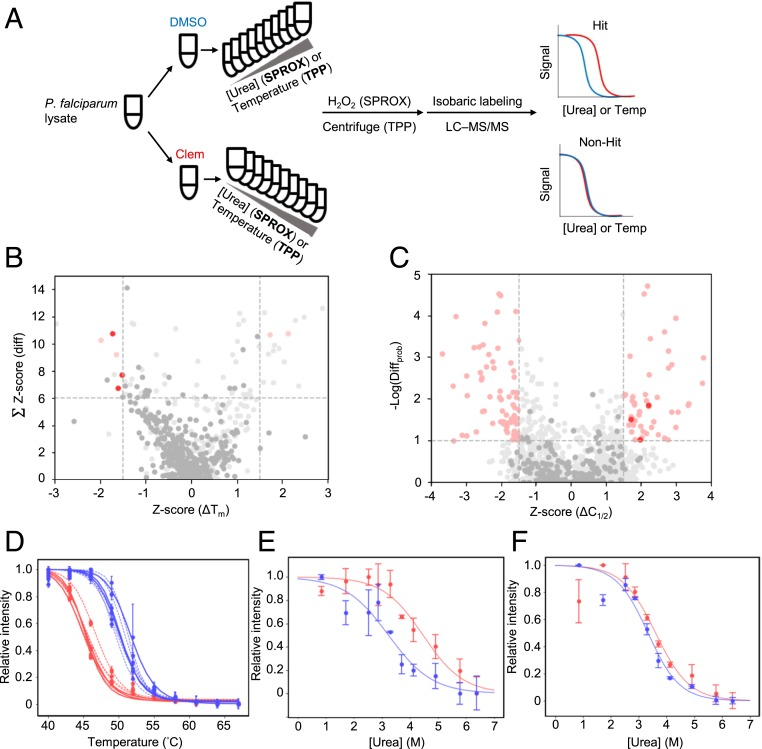
Figure 7. SPROX Identification of Drug Targets [7]
4.Characterization of the Saccharomyces Cerevisiae ATP-Interactome Using the iTRAQ-SPROX Technique
The SPROX technique has been combined with isobaric mass labeling strategies to identify adenosine triphosphate (ATP)-interacting proteins in the Saccharomyces cerevisiae proteome. The SPROX method used in this work enabled the determination of ATP interactions (both direct and indirect) of 373 proteins in yeast cell lysates using the non-hydrolyzable ATP analog, adenyl imidodiphosphate (AMP-PNP). A total of 28 proteins were identified with AMP-PNP-induced thermodynamic stability changes. These protein hits included 14 proteins that were previously annotated as ATP-binding proteins in the Saccharomyces Genome Database (SGD). The 14 unannotated ATP-binding proteins included 9 proteins that were previously found to be ATP-sensitive in earlier SPROX studies using SILAC-based approach. Bioinformatics analysis of protein hits found in this paper and earlier SILAC-SPROX experiments had shown that many of the previously annotated ATP-binding protein hits were kinases, ligases, and chaperones. In contrast, many of the newly discovered ATP-sensitive proteins were not from these protein classes, but rather were from hydrolases, oxidoreductases, and nucleic acid-binding proteins.
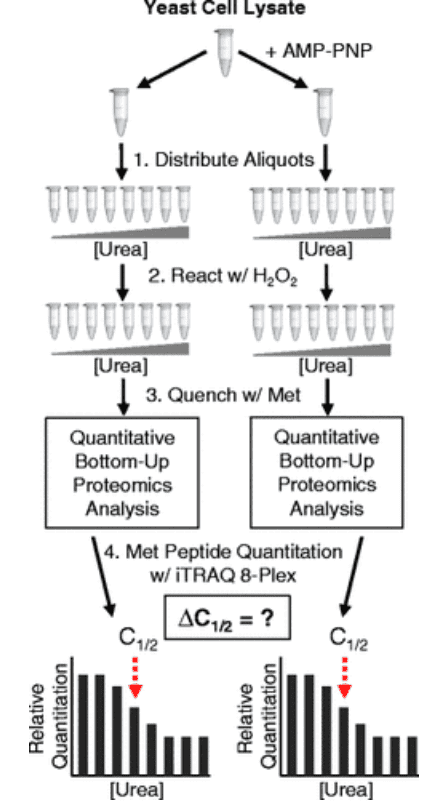
Figure 8. The iTRAQ-SPROX Technique Characterizes the ATP-Interactome of Saccharomyces Cerevisiae [8]
Sample Submission Requirements
1. Samples such as drugs/proteins should not be contaminated with impurities as much as possible.
Services at MtoZ Biolabs
1. The Complete Experimental Procedure
2. Relevant Instrument Parameters
3. Raw MS Data
4. Data Analysis Reports
Applications
1.Natural Product Target Identification and Validation
The identification of natural product target proteins is critical to understanding the mechanism of action of natural products developed for use as molecular probes and potential therapeutics. Affinity chromatography (AC) of immobilized natural products has been routinely used to identify target proteins with good results. However, this method has limitations, as labeling or labeling for immobilization and affinity purification often result in reduction or alteration to activity of the natural product. Novel strategies have recently been developed and applied to identify natural products and synthesize small molecule target proteins without chemical modification of natural products. These direct and indirect methods for the identification of label-free natural product targets include DARTS, SPROX, thermal shift assay, MS-based thermal stability profiling, and bioinformatics-based connectivity analysis. The integration of newly developed technologies will provide new insights and highlight the value of natural products as bioprobes and new drug candidates.
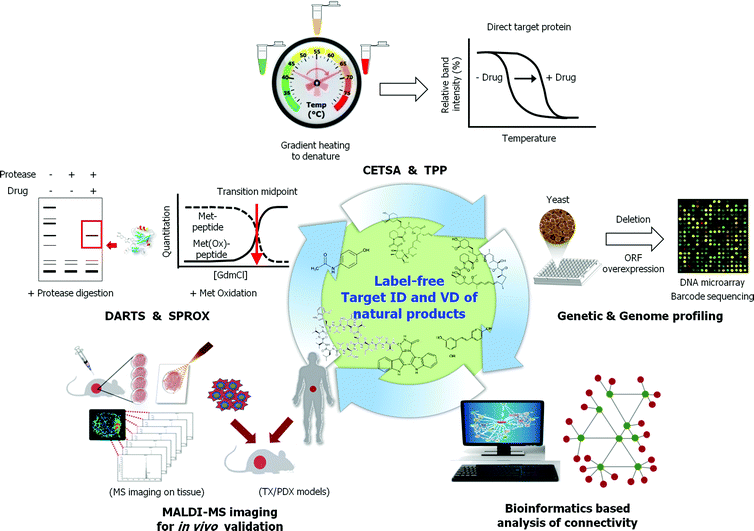
Figure 9. Use Label-Free Methods for Natural Product Target Identification and Validation [1]
FAQ
Q1: What are the advantages and disadvantages of SPROX?
Although SPROX has many advantages over MS-based thermal stability profiling and DARTS, it also has some limitations. First, since methionine oxidation is a measurement parameter, SPROX analysis can only be used for peptides containing methionine. Second, not all methionine residues exhibit different oxidation rates. Third, detailed information on the ligand binding site is not available. Finally, it cannot be applied to living cells due to the need for some degree of chemical denaturation and the addition of oxidants. Therefore, SPROX can be used to determine direct and indirect protein targets for drug molecules, as well as to determine the presence of protein folding and predict potential side effects of small molecule drugs.
References
[1] Chang J, Kim Y, Kwon HJ. Advances in identification and validation of protein targets of natural products without chemical modification. Nat Prod Rep. 2016 May 4;33(5):719-30. doi: 10.1039/c5np00107b. PMID: 26964663.
[2] West GM, Tucker CL, Xu T, Park SK, Han X, Yates JR 3rd, Fitzgerald MC. Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc Natl Acad Sci U S A. 2010 May 18;107(20):9078-82. doi: 10.1073/pnas.1000148107. Epub 2010 May 3. PMID: 20439767; PMCID: PMC2889096.
[3] Kaur U, Meng H, Lui F, Ma R, Ogburn RN, Johnson JHR, Fitzgerald MC, Jones LM. Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale. J Proteome Res. 2018 Nov 2;17(11):3614-3627. doi: 10.1021/acs.jproteome.8b00341. Epub 2018 Oct 8. PMID: 30222357; PMCID: PMC6524533.
[4] Geer MA, Fitzgerald MC. Energetics-based methods for protein folding and stability measurements. Annu Rev Anal Chem (Palo Alto Calif). 2014;7:209-28. doi: 10.1146/annurev-anchem-071213-020024. Epub 2014 May 28. PMID: 24896313.
[5] Ma R, Johnson JHR, Tang Y, Fitzgerald MC. Analysis of Brain Protein Stability Changes in Mouse Models of Normal Aging and α-Synucleinopathy Reveals Age- and Disease-Related Differences. J Proteome Res. 2021 Nov 5;20(11):5156-5168. doi: 10.1021/acs.jproteome.1c00653. Epub 2021 Oct 4. PMID: 34606284; PMCID: PMC9288659.
[6] Liu F, Meng H, Fitzgerald MC. Large-Scale Analysis of Breast Cancer-Related Conformational Changes in Proteins Using SILAC-SPROX. J Proteome Res. 2017 Sep 1;16(9):3277-3286. doi: 10.1021/acs.jproteome.7b00283. Epub 2017 Jul 27. PMID: 28673085; PMCID: PMC5605765.
[7] Lu KY, Quan B, Sylvester K, Srivastava T, Fitzgerald MC, Derbyshire ER. Plasmodium chaperonin TRiC/CCT identified as a target of the antihistamine clemastine using parallel chemoproteomic strategy. Proc Natl Acad Sci U S A. 2020 Mar 17;117(11):5810-5817. doi: 10.1073/pnas.1913525117. Epub 2020 Mar 3. PMID: 32127489; PMCID: PMC7084109.
[8] Geer MA, Fitzgerald MC. Characterization of the Saccharomyces cerevisiae ATP-Interactome using the iTRAQ-SPROX Technique. J Am Soc Mass Spectrom. 2016 Feb;27(2):233-43. doi: 10.1007/s13361-015-1290-z. Epub 2015 Nov 3. PMID: 26530046.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

