





Reference-Based Transcriptome Sequencing Service
Transcriptome sequencing has become an essential tool for exploring gene expression, identifying novel transcripts, and studying transcriptomic complexity across different biological conditions. Reference-based transcriptome sequencing is a high-resolution RNA-Seq strategy that leverages pre-existing genome assemblies to accurately map transcriptomic reads and reconstruct the transcriptome. Unlike de novo transcriptome assembly, which infers transcript structures solely from raw sequencing reads, the reference-based approach uses the reference genome as a scaffold, enabling more precise identification of exon-intron boundaries, alternative splicing events, and low-abundance transcripts.
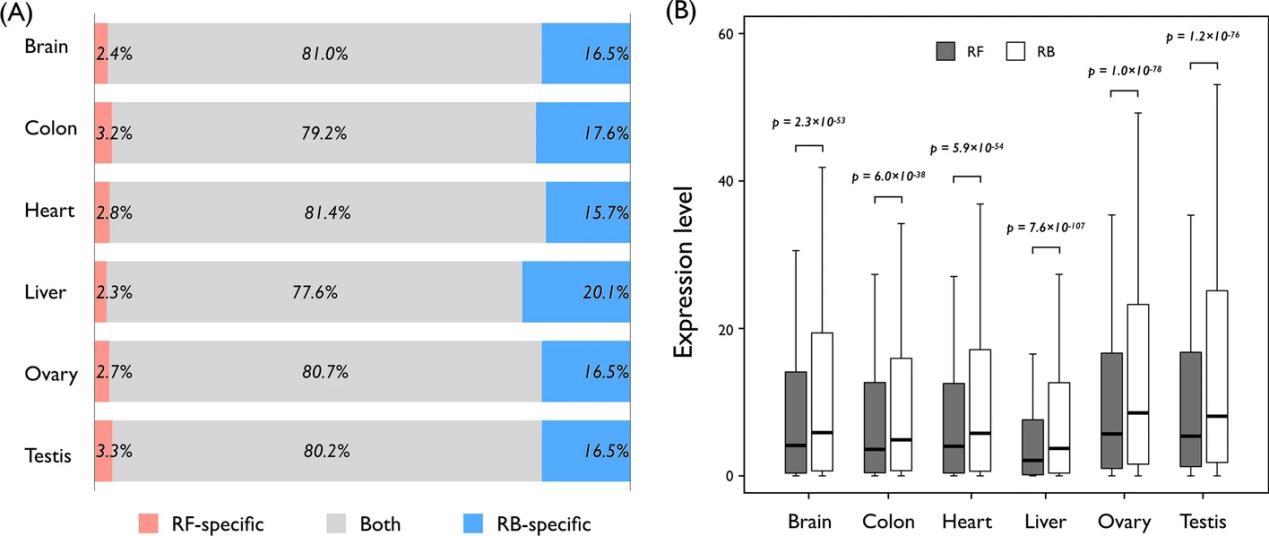
Lee, SG. et al. BMC Bioinformatics. 2021.
Figure1. Comparison of Transcriptome Profiles Between Reference-Free (RF)
and Reference-Based (RB) Methods
Reference-based transcriptome sequencing is particularly beneficial for organisms with well-annotated genomes, such as Homo sapiens, Mus musculus, Arabidopsis thaliana, and various sequenced non-model species. It supports a wide range of applications including transcript quantification, gene expression analysis, novel transcript discovery, and the characterization of alternative splicing events.
Service at MtoZ Biolabs
MtoZ Biolabs offers a comprehensive and professional reference-based transcriptome sequencing service that covers every step from RNA extraction and library preparation to sequencing and bioinformatics analysis. Powered by high-throughput Illumina platforms and a rigorous multi-level quality control system, we deliver high-precision and scalable transcriptome solutions tailored to various research depths and sample sizes.
Depending on the research objectives, we provide a range of library construction strategies, including poly(A) enrichment for mRNA analysis, rRNA depletion for studies involving lncRNA, circRNA, or whole transcriptome profiling, and strand-specific library preparation to enhance isoform resolution. Our services support a wide array of model organisms such as human, mouse, rat, zebrafish, Arabidopsis thaliana, rice, wheat, and corn. Whether you are investigating disease mechanisms, drug response pathways, tissue- or stage-specific gene expression, or engaging in integrative multi-omics research, MtoZ Biolabs is committed to delivering stable, reproducible, and publication-ready transcriptome data to support your scientific goals.
Analysis Workflow
1. Sample Preparation
Evaluate RNA integrity (RIN), quantify concentration, and assess degradation. Accepts tissues, cells, or purified RNA samples.
2. Library Construction
Select from poly(A) enrichment, rRNA depletion, or strand-specific library prep. Construct paired-end cDNA libraries with 250–300 bp inserts.
3. High-Throughput Sequencing
Perform paired-end 150 bp (PE150) sequencing on the NovaSeq 6000 platform. Standard output: ≥20 million reads per sample or ≥10 Gb clean data.
4. Data Analysis
Align reads using HISAT2 and quantify expression with StringTie. Conduct differential expression analysis via DESeq2 or edgeR with functional annotation and visualized results.
Why Choose MtoZ Biolabs?
✅High-Throughput Sequencing Platform
Utilizing Illumina's high-throughput sequencing platforms ensures high coverage and low error rates, meeting the demands of in-depth expression profiling and alternative splicing analysis.
✅Broad Species Compatibility
Compatible with a wide range of model organisms including human, mouse, Arabidopsis thaliana, and zebrafish, as well as non-model species with available reference genomes.
✅Comprehensive Transcript Coverage
Captures a broad spectrum of transcript types, including mRNAs, lncRNAs, circRNAs, novel isoforms, and previously unannotated transcripts.
✅One-Time-Charge
Our pricing is transparent, no hidden feesor additional costs.
✅High-Data-Quality
Deep data coverage with strict data qualitycontrol. AI-powered bioinformatics platform integrates all reference based transcriptome sequencing data, providing clients with a comprehensive data report.
Application Examples
1.Differential Expression Analysis in Disease: Identifying Key Pathogenic Genes
Reference-based transcriptome sequencing was applied to synovial tissues from rheumatoid arthritis patients, revealing several upregulated genes in the TNF signaling pathway that play key roles in amplifying inflammation.(Zhou Y. et al., 2021, Nature Communications)
2.Splice Isoform Profiling: Characterizing Cancer-Related Alternative Splicing Events
Transcriptome sequencing of breast cancer tissues enabled the systematic identification of prognostic alternative splicing events and facilitated the development of a predictive splicing model.(Sebestyén E. et al., 2020, Genome Biology)
3.Non-Coding RNA Research: Identifying Functional lncRNAs
A reference-based strategy was used to detect dynamically expressed lncRNAs during neuronal development, and several were validated for their regulatory roles in axonal growth.(Li J. et al., 2022, Cell Reports)
4.Agricultural Research: Dissecting Plant Stress Response Mechanisms
In a study on salt stress tolerance in rice, transcriptome sequencing revealed key signaling pathways and regulatory genes, advancing research in salt-tolerant crop breeding.(Wang L. et al., 2021, Plant Physiology)
5.Transcript Re-annotation: Enhancing Reference Genome Annotation
Sequencing of zebrafish embryos at different developmental stages led to the construction of a stage-specific transcript database, significantly improving existing genome annotations.(Sun Y. et al., 2023, BMC Genomics)
Sample Submission Suggestions

Storage and Shipping Guidelines: To preserve RNA integrity, avoid repeated freeze-thaw cycles. Total RNA should be stored in RNase-free tubes at −80℃ and shipped on dry ice. For cell and tissue samples, rapid freezing in liquid nitrogen is recommended prior to shipping.
Note: If your study focuses on lncRNA or circRNA analysis, please notify us in advance so that we can select the appropriate rRNA depletion or library construction strategy.
For any special sample types or handling concerns, feel free to contact our technical support team for customized sample preparation guidance.
Deliverables
1. Raw sequencing data in FASTQ format
2. Alignment files in BAM format
3. Gene expression quantification matrix (TPM, FPKM, Counts)
4. Differential expression analysis results (including gene ID, log₂FC, p-value, adjusted p-value)
5. Functional enrichment analysis results (GO/KEGG enrichment tables)
Related Service
Transcriptome Sequencing (RNA-sequencing) Service
Metatranscriptomics Sequencing Service
Full-Length 16S/18S/ITS Sequencing Service
Prokaryotic Transcriptomics Sequencing Service
How to order?

