





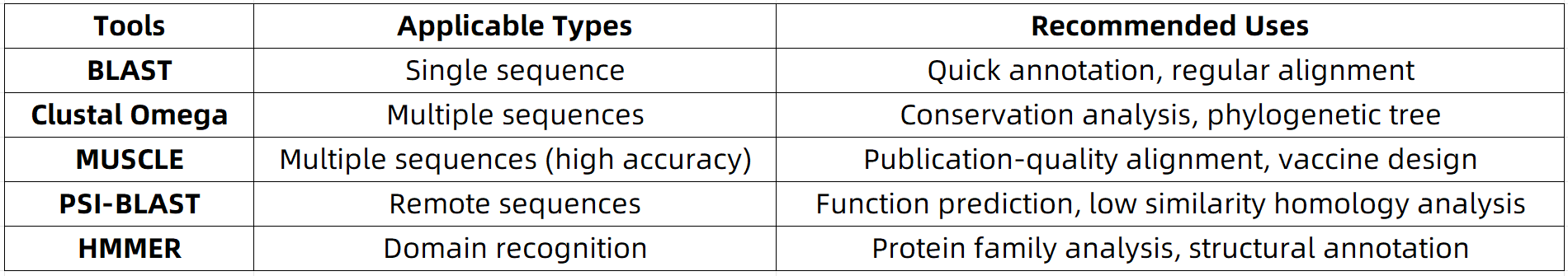
Recommendations for 5 Commonly Used Protein Sequence Alignment Tools
-
Supports various alignment modes such as protein-to-protein (blastp) and nucleotide-to-protein (blastx)
-
Integrated with the comprehensive NCBI database, covering a broad range of species
-
Enables offline analysis using user-defined custom databases
-
Quickly infer the potential function or species origin of unknown proteins
-
Evaluate the functional conservation of mutation sites associated with disease
-
Capable of aligning dozens to hundreds of sequences efficiently
-
Outputs formats that are compatible with phylogenetic tree construction
-
Offers significantly improved performance compared to earlier versions like ClustalW
-
Investigating evolutionary relationships within protein families
-
Mapping the distribution of conserved domains and functional motifs
-
Produces high-fidelity alignments, ideal for closely related sequences
-
Compatible with various visualization tools such as Jalview
-
Analysis of sequence variation among viral or bacterial strains
-
Subtype-specific protein analysis and identification of vaccine target candidates
-
Generates position-specific scoring matrices (PSSMs)
-
Particularly effective for evolutionary analysis and functional inference
-
Functional prediction of poorly annotated or unknown proteins
-
Investigating functional conservation across distantly related species
-
Supports the Pfam database and excels in protein domain identification
-
Offers higher precision than conventional alignment tools, especially for short sequences
-
Annotation of conserved domains and classification of protein families
-
Proteomics database searches and functional annotation of proteins
-
Target Screening: Integration of BLAST and HMMER enables precise identification of disease-associated proteins and their functional families.
-
Functional Annotation: Combined use of Clustal Omega and Pfam facilitates high-quality multiple sequence alignment and domain annotation.
-
Mutation Analysis: PSI-BLAST, in conjunction with structure prediction tools such as AlphaFold, is used to assess the functional impact of mutations.
-
High-Throughput Data Mining: By integrating mass spectrometry-based quantification with sequence-level analysis, target prioritization models are constructed.
Introduction: Why Is Protein Sequence Alignment so Important?
The function of a protein is often reflected in its amino acid sequence. Conserved regions, active sites, and modification sites within the sequence serve as critical indicators for predicting protein function. By aligning a target sequence with known protein sequences in public databases, researchers can: annotate the functions of uncharacterized proteins; identify homologous proteins and evolutionary relationships; detect domains, binding sites, or antigenic epitopes; and support downstream tasks such as protein structure prediction, molecular docking, and drug screening. Selecting an appropriate alignment tool not only improves the accuracy of the results but also significantly enhances the efficiency of the analysis.
Protein Sequence Alignment Tools
1. BLAST (Basic Local Alignment Search Tool)
As one of the most established and widely used tools for sequence alignment, BLAST is often the first step in protein analysis. It performs local alignments to rapidly identify segments of the query sequence that are similar to those found in reference databases.
(1) Use: Rapid local sequence alignment and identification of homologous proteins
(2) Website: https://blast.ncbi.nlm.nih.gov
(3) Highlights:
(4) Suitable scenarios:
2. Clustal Omega
Clustal Omega is widely regarded as a standard tool for multiple sequence alignment, particularly suitable for conducting evolutionary analyses and identifying conserved functional regions across a group of protein sequences.
(1) Use: Multiple sequence alignment for detecting conserved regions
(2) Website: https://www.ebi.ac.uk/Tools/msa/clustalo/
(3) Highlights:
(4) Suitable scenarios:
3. MUSCLE
MUSCLE is similar to Clustal Omega in functionality but places a greater emphasis on alignment accuracy, making it particularly suitable for constructing phylogenetic trees used in peer-reviewed research.
(1) Use: High-accuracy multiple sequence alignment
(2) Website: https://www.ebi.ac.uk/Tools/msa/muscle/
(3) Highlights:
(4) Suitable scenarios:
4. PSI-BLAST (Position-Specific Iterated BLAST)
PSI-BLAST extends the standard BLAST algorithm by iteratively refining a position-specific scoring matrix (PSSM), making it suitable for detecting distantly related yet functionally relevant proteins that may not be captured by standard BLAST searches.
(1) Purpose: Detection of remote homologous proteins
(2) Website: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins
(3) Highlights:
(4) Application Scenarios:
5. HMMER
HMMER performs protein sequence alignment based on hidden Markov models, with particular strength in identifying conserved domains and sequence motifs characteristic of protein families.
(1) Purpose: Sequence alignment using hidden Markov models
(2) Website: http://hmmer.org/
(3) Highlights:
(4) Application Scenarios:
Summary and Recommendations
MtoZ Biolabs: A One-Stop Platform for Protein Sequence Alignment and Annotation
MtoZ Biolabs integrates a suite of alignment algorithms into an automated, multi-layered protein annotation pipeline that supports various stages of proteomics and drug discovery projects. Its applications include:
For projects involving unknown protein annotation, target discovery, or functional site prediction, MtoZ Biolabs offers a comprehensive, end-to-end solution—from raw data preprocessing to the generation of visualized reports—designed to accelerate your research progress.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

