





Protein Identification Service by Tandem Mass Spectrometry
Protein tandem mass spectrometry (MS/MS) is an efficient, precise, and sensitive method for qualitative and quantitative identification of proteins based on mass spectrometry technology. It is widely used in proteomics research, biopharmaceutical quality control, disease biomarker discovery, and other fields. By analyzing the peptide fragments (products of protein digestion) using mass spectrometry, this technique identifies target proteins, including unknown ones. Originating in the early 1990s, it was initially dominated by liquid chromatography coupled with mass spectrometry (LC-MS/MS). With the development of techniques such as matrix-assisted laser desorption/ionization (MALDI), protein mass spectrometry identification entered a new era. Today, MS/MS has become an important tool in proteomics research, with significant implications for deciphering protein structures and functions in complex biological samples. It aids in uncovering protein interactions, signal transduction mechanisms, and drives advancements in biological science and disease treatment.
Analysis Workflow
1. Sample Preparation
Purify, concentrate, and desalt the provided protein samples from customers using appropriate methods such as high-performance liquid chromatography (HPLC) or gel electrophoresis to remove impurities and enhance protein purity.
2. Protein Digestion
Perform multiple enzymatic digestions on purified proteins to cleave them into peptide fragments, facilitating subsequent mass spectrometry analysis.
3. Peptide Separation
Separate peptide fragments using nano-high-performance liquid chromatography (nano-HPLC) to enhance resolution and detection sensitivity effectively.
4. MS/MS Analysis
Employ high-resolution, high-sensitivity mass spectrometers (Thermo Fisher's Orbitrap Fusion Lumos, Orbitrap Exploris 240) to analyze the quality and fragment ions of separated peptide fragments through MS/MS.
5. Data Analysis
Utilize specialized data processing software such as Mascot and protein databases for peptide quality matching and fragment ion searching, ultimately achieving protein identification, including detailed information such as protein name, amino acid sequence, and matching score.
Applications
1. High-Throughput Protein Identification
MS/MS plays a critical role in proteomics research, identifying and quantifying proteins in biological samples, revealing differences in protein expression, interactions, and functions.
2. Post-translational Modification Studies
Used for identifying post-translational modifications of proteins (such as phosphorylation, acetylation, glycosylation), revealing the impact of modifications on protein function and signaling.
3. Protein Interaction Studies
By identifying components of protein complexes, it reveals the interaction relationships between proteins, providing a basis for the study of biological signaling pathways.
4. Protein Structure Analysis
MS/MS technology can analyze the primary structure (amino acid sequence) and some secondary and tertiary structure information of proteins, providing reference data for protein structure research.
5. Disease Diagnosis and Biomarker Research
MS/MS technology can be used for the identification and quantification of disease-related proteins, providing a basis for disease diagnosis and prognosis assessment.
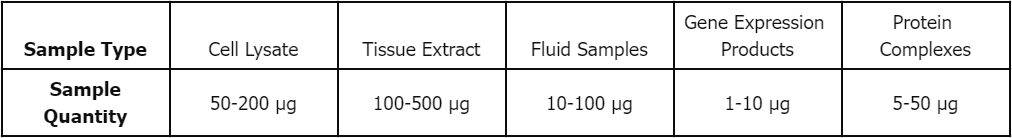
Sample Submission Requirements
Deliverables
In the technical report, MtoZ Biolabs will provide you with a detailed report including:
1. Experimental Procedures
2. Relevant Mass Spectrometry Parameters
3. Detailed Information on Protein Tandem Mass Spectrometry Identification
4. Mass Spectrometry Images
5. Raw Data
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Mass Spectrometry-Based Protein Identification Service
How to order?

