





Degrader Design and Synthesis | PROTAC Service
PROTACs represent a novel strategy for protein degradation that has gained prominence recently. Utilizing bifunctional molecules, PROTACs enable the targeted ubiquitination and subsequent degradation of proteins via the UPS. PROTACs can not only offer potential therapeutic applications for a variety of conditions, including cancer, immune dysregulation, viral infections, and neurodegenerative diseases, but also provide a unique chemical knockdown tool for biological research in a catalytic, reversible, and rapid manner.
A PROTAC molecule comprises two functional parts: an E3 ligase recruiter and a ligand for the protein of interest (POI). By bringing the E3 ligase and POI into proximity, PROTACs initiate the process of ubiquitination, leading to the degradation of the target protein by the proteasome. This interaction enables the transfer of ubiquitin from the E3 ligase to the POI, marking it for recognition and breakdown into peptides by the proteasome. Both the E3 ligase and the target protein can bind simultaneously to a single PROTAC molecule. Through the orchestrated actions of E1, E2, and E3 ligases, ubiquitin is repeatedly transferred to the target, facilitating its polyubiquitination and subsequent degradation.
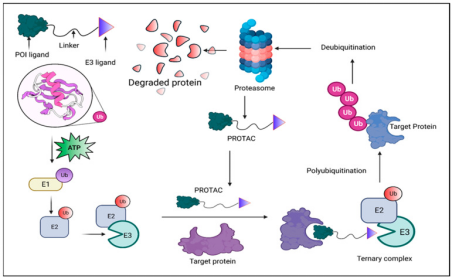
Figure 1. Principle of PROTAC-Mediated Degradation [4]
PROTAC technology shows vast potential across a range of diseases, demonstrating significant efficacy not only in oncology but also in managing immune disorders, viral infections, and neurodegenerative conditions. It can regulate growth and developmental processes through key signaling pathways and proteins, such as PI3K/AKT and RAS-RAF-MEK-ERK pathways and the p53 protein. Both conventional and unconventional PROTACs can target these critical elements involved in drug development.
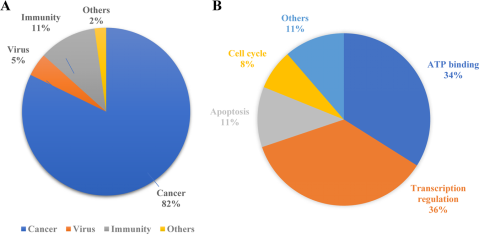
Figure 2. Applications of PROTAC [5]
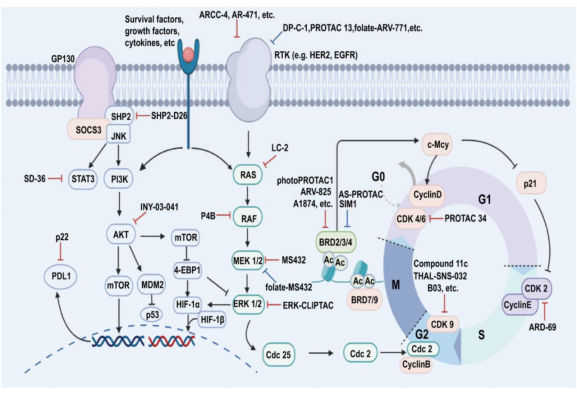
Figure 3. Representative PROTACs in Drug Development [6]
According to previous reports, researchers tend to choose kinases as targets for protein degradation. Incomplete statistics show that different degraders based on PROTAC technology can degrade about 54 kinases, accounting for 45% of the total targets. This preference is largely due to the availability of effective kinase inhibitors or ligands, which can be readily adapted for PROTAC development, ensuring strong binding affinity. Additionally, the deep binding pockets of kinases facilitate PROTAC binding, enhancing the ubiquitination and degradation process. Despite the high homology among kinase proteins, PROTACs can selectively target different kinase isoforms, even those originally identified by non-selective inhibitors.
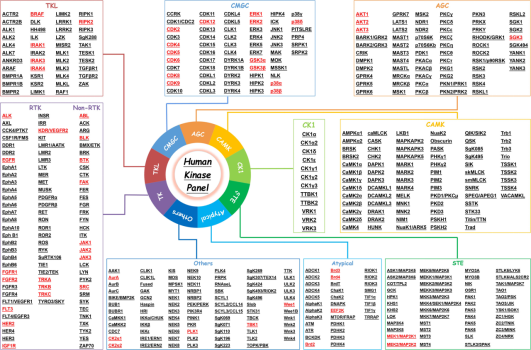
Figure 4. List of PROTAC-Based Human "Degradable" Kinases [7]
The rapid advancement of PROTAC technology offers new directions for biopharmaceutical research, with benefits including the inhibition of tumor growth, overcoming drug resistance, targeting multiple pathways, and reducing adverse effects. However, challenges such as high molecular weight, spatial hindrance from short linkers, incompatibility of linkers with specific E3 ligases, and the selection of appropriate target recruitment ligands remain. Early PROTAC design should consider the ligand for the target protein, the E3 ligase ligand, and the linker, optimizing these components to address the challenges posed by PROTAC's molecular size. Innovations in artificial intelligence, virtual screening, cryo-electron microscopy, and X-ray crystallography could usher in a new era of structure-based PROTAC design, offering insights into efficient molecular design for ligand selection, optimal linker strategies, and the selective targeting of POI and E3 ligase pairs. These advances hold the promise of enhancing the selectivity, therapeutic efficacy, and drug-like properties of PROTAC molecules.
The PROTAC molecule consists of three parts: a target protein ligand (a small molecule that binds to a POI and is primarily used to target and capture the POI), an E3 ubiquitin ligase ligand (a small molecule that binds to a ubiquitin ligase and is responsible for the specific recruitment of E3 ubiquitin ligase), and a linker (a link between the first two that binds two ligands to form a stable ternary complex).

Figure 5. Types of PROTACs [8]
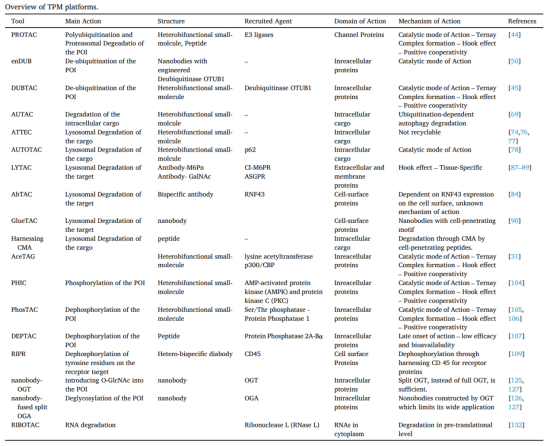
Figure 6. Characteristics of Different Types of PROTACs [9]
1. Target Protein Ligand
As the guiding group of PROTAC, the target protein ligand is responsible for capturing the target protein and thus needs to have high coordination capability and selectivity. In practical applications, existing agonists of a protein can be used as ligand candidates, and for proteins without reported molecules, high-throughput screening and virtual screening can be employed to find ligands. It is not required that the ligand binds to the protein in an inhibitory or competitive manner, but it must have high specificity for the target protein.
When selecting a target protein, consider:
(1) Specificity of the protein, as non-specific binding can lead to off-target effects.
(2) Whether the protein is typically degraded by PROTAC molecules.
(3) Availability of related ligands or crystal structures, which are vital for efficient PROTAC design.
A conventional PROTAC design can proceed if these conditions are met, leveraging known ligands’ structure-activity relationships to identify modifiable positions or using protein crystal structures for more targeted virtual screening.
2. E3 Ligase Ligand
The CRBN and von Hippel-Lindau (VHL) ligands are predominantly used for recruiting E3 ligases. Selection of the appropriate E3 ligase ligand typically involves assessing the abundance of specific ligases in target cells using assay kits to ensure optimal interaction.
Table 1. E3 Ligases Recruited for PROTAC Construction: Characteristics and Limitations [10]
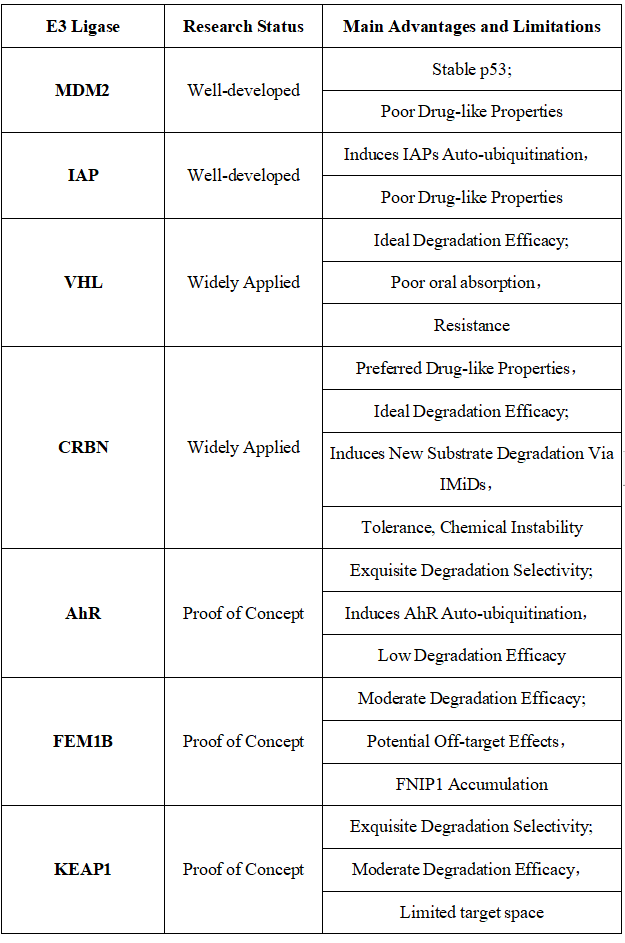
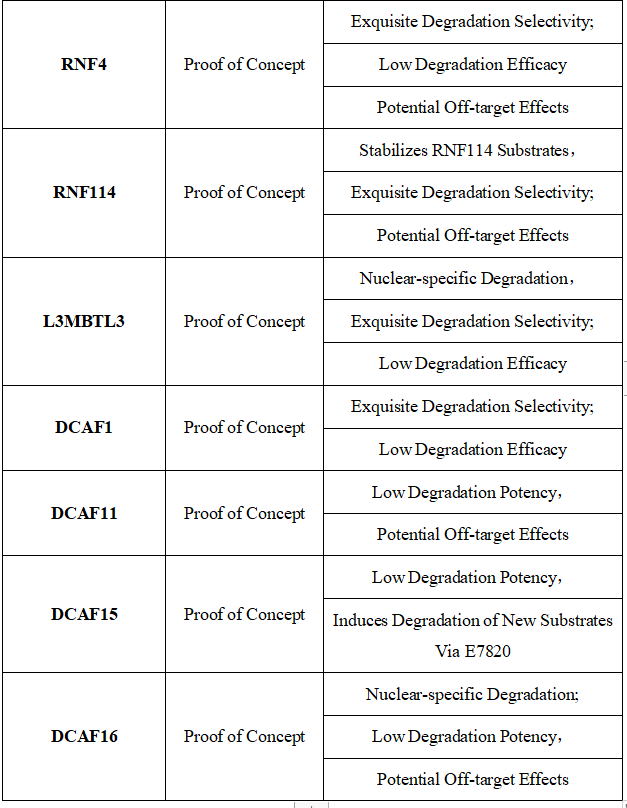
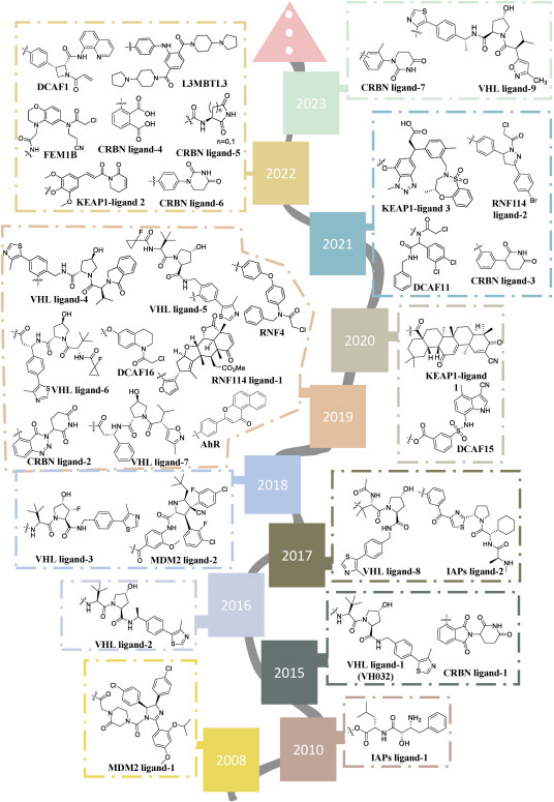
Figure 7. Timeline of E3 Ligase Ligand Identification in PROTAC Design [10]
3. Linker
The linker plays a decisive role in the physicochemical properties and biological activity of PROTAC compounds. The correct combination of the linker's length, hydrophilicity, and rigidity forms the basis for successfully designing effective PROTACs. Various linkers are usually based on polyethylene glycols (PEGs), unsaturated alkane chains, and triazoles. They are typically connected to ligands via amines, amides, ethers, alkylamines, and single and multiple C-C bonds. Most linkers contain a combination of hydrophobic (unsaturated alkanes) and hydrophilic parts (PEGs, piperidines, amides, triazoles, etc.), to balance the resulting compound. Initial PROTAC designs often choose PEG chains as the linking molecule due to their flexibility, which allows for adjustment of the molecule to the appropriate binding angle. However, the flexibility of PEG chains also introduces high binding entropy, which needs to be improved in later optimization stages.
When designing effective PROTACs, one of the most critical decisions is choosing the appropriate linker length to connect the ligands binding the POI and E3 ligase. The composition and length of the linker play a crucial role in the efficacy and physicochemical properties of the PROTAC compounds. If the linker is too short, the two ligands cannot simultaneously bind to their respective proteins due to steric clashes resulting in failure of ternary complex formation. On the other hand, if the linker is too long then two proteins cannot be brought in close proximity with each other for target ubiquitination. Currently, structural optimization of PROTACs mainly focuses on the evaluation of structure-activity relationship (SAR) for various size linkers. Typically, evaluations start with longer version of linkers, gradually reducing the length until the activity is lost. Most linkers used have a chain length of 10 to 20 atoms. Additionally, studies have summarized the relationship between chain length and degradation activity through normalization analysis. Longer chain lengths reduce activity due to higher entropy, but this effect is minor compared to the inability of shorter lengths to effectively bring the ends of the ligand molecules closer together, hence the initial molecular design generally opts for a slightly longer linker chain.
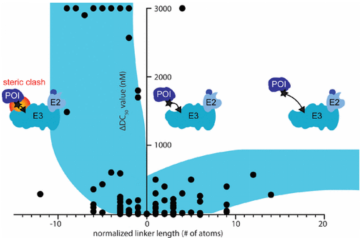
Figure 8. Relationship Between Linker Chain Length and Activity Under Normalization [11]
4. Assembly of PROTACs
In the assembly of PROTACs, taking VHL as an example, during the synthesis of VHL-targeting PROTACs, the chemical type of the linker (flexible or rigid) is predetermined by the functionality of the VHL ligand and POI ligand. Traditionally, there are three methods for assembling VHL-based PROTACs: (a) First coupling the linker to the VHL ligand, then connecting it to the warhead; (b) First tethering the linker to the warhead, then coupling it with the VHL ligand; (c) Installing two smaller linker fragments on both the POI and E3 ligands and connecting the halves. In all methods, the linkage point (R) on the warhead is often modified to facilitate conjugation. The choice of method is usually based on two factors: (1) the availability and cost of building blocks, (2) the number of steps required to synthesize the warhead and linkers.
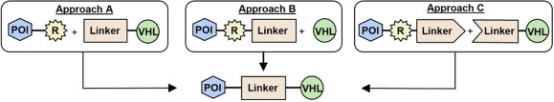
Figure 9. Schematic of Assembling VHL-Based PROTACs [12]
Analysis Workflow
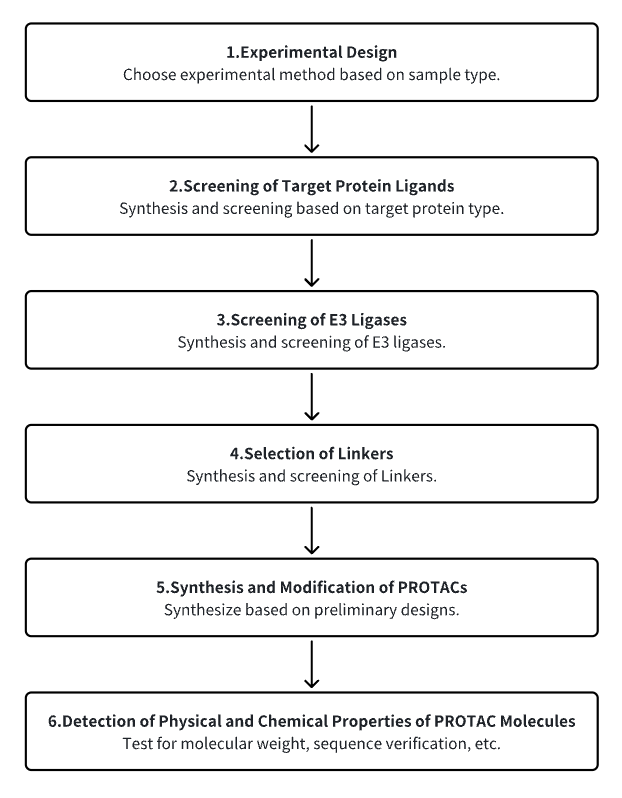
1. Establish the Experimental Workflow Based on Specific Research Needs
2. Screen for Target Protein Ligands
3. Select E3 Ubiquitin Ligase Ligands
4. Choose Appropriate Linker Molecules
5. Synthesize and Modify PROTACs
6. Detect the Physicochemical Properties of PROTAC Molecules
Service Advantages
1. Provide Professional PROTAC Design Consultation to Facilitate Efficient and Rational Drug Design
2. Offer Rapid, Cost-Effective, and High-Quality Synthesis Services
3. Make Available a Commercial Library of PROTACs for Selection
Sample Results
1. Selective Targeting of Protein Degradation Using DNA Framework-Designed Chimeric Platforms
One challenge in developing PROTACs is to establish a universal platform suitable for various scenarios to precisely degrade POIs. Inspired by the addressability, programmability, and structural rigidity of DNA frameworks, a new study has introduced covalently linked DNA-framework-based PROTACs (DbTACs), which can be synthesized in high-throughput via facile bioorthogonal chemistry and self-assembly. Utilizing DNA tetrahedra as templates, this approach precisely defines the spatial arrangement of ligands, allowing control over ligand distances from 8 Å to 57 Å. Research indicates that DbTACs configured with optimal linker lengths between ligands achieve higher degradation rates and enhanced binding affinity. Bispecific DbTACs (bis-DbTACs) with trivalent ligand assembly enable multi-target depletion while maintaining highly selective degradation of protein subtypes. DbTACs have shown potent efficacy in degrading diverse targets, including protein kinases and transcription factors across different cellular compartments, when equipped with various types of warheads (small molecules, antibodies, and DNA motifs). Overall, this modular DNA framework approach offers a broad platform that not only enhances understanding of degradation agent design principles but also provides promising avenues for guiding drug discovery.
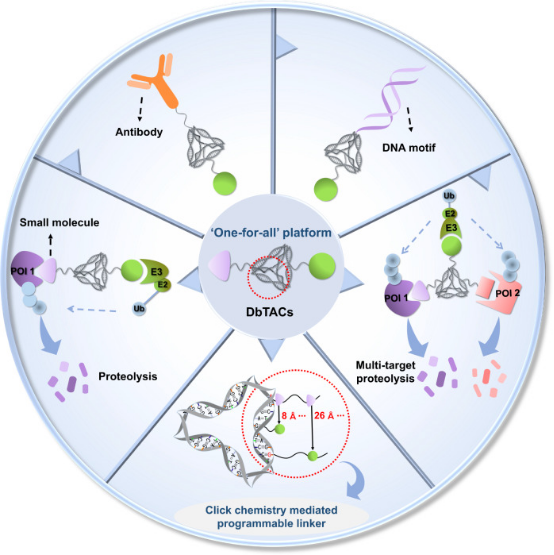
Figure 10. DbTACs Platform Designed for Selective Targeted Protein Degradation [13]
2. Development of a DCAF4-Based CDK6/16 PROTAC
Significant advances have been made in breast cancer treatment over recent decades, yet triple-negative breast cancer (TNBC), known for its high malignancy, remains challenging to treat effectively. The abnormal cell cycle, prevalent in human breast cancer and other malignancies, positions cyclin-dependent kinase (CDK) 4/6 as a critical regulatory target. Leveraging the predominantly nuclear localization of the DCAF16 E3 ligase, researchers have crafted a series of DCAF16-based CDK4/6 degraders by combining Palbociclib with the DCAF16 E3 ligase ligand KB02 and various linkers. Notably, compound A4 has shown potent inhibitory effects on CDK4/6, significantly reducing CDK6/231 protein levels in MDA-MB-4 cells in a dose- and time-dependent manner. Moreover, A4 exhibited considerably lower toxicity in normal cells compared to Palbociclib and demonstrated therapeutic potential in an in vivo MDA-MB-231 xenograft model, suggesting its viability as a novel DCAF4-based CDK6/16 degrader for further research in TNBC treatment.
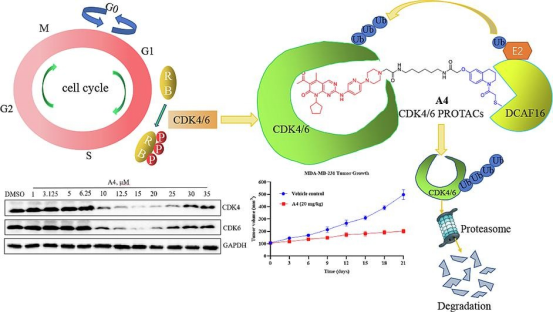
Figure 11. PROTAC Targeting CDK4/6 [14]
3. UGI Reaction-Assisted Assembly of Covalent PROTACs Targeting Glutathione Peroxidase 4 (GPX4)
Inactivating GPX4 to induce cell ferroptosis is an emerging strategy in cancer therapy. Despite this, only a few inhibitors of GPX4 are currently available. PROTAC technology offers a promising alternative to traditional therapies, overcoming some of their limitations. This paper describes the assembly of the PROTAC-like activity-based probe PD-Q2 using the Ugi reaction, which combines a known GPX4 inhibitor ML-162 homolog to the E3 ligase cereblon ligand-pomalidomide. Pull-down and immunoblot analyses confirm that GPX4 was a covalent target of PD-Q2, but the degradation efficiency was weak. Therefore, a series of degraders was further synthesized by varying the linkers of heterofunctional PROTACs. Among these, PD-4 and PD-P2 were found to efficiently degrade GPX4 via the UPS, inducing lipid ROS accumulation and effectively inhibiting colony formation and cell growth. Notably, when using pomalidomide, the degraders displayed high fluorescence signals, predominantly localized within lysosomes, which may impact their anti-cell proliferation efficacy. Collectively, these findings contribute to the development of GPX4 degraders and further explore their potential in modulating ferroptosis for therapeutic purposes.
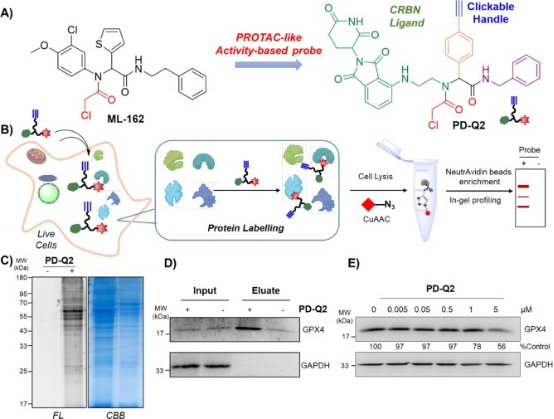
Figure 12. Design of the PROTAC-like Activity Probe PD-Q162 for GPX2 Inhibition (ML-4) [15]
Sample Submission Requirements
1. Comprehensive Experimental Steps
2. Specifications of Relevant Instrumentation
3. Compilation of Original Experimental Data
4. Detailed Reports on PROTAC Synthesis and Analysis
Applications
1. PROTACs as New Tools for Enhancing Cancer Immunotherapy
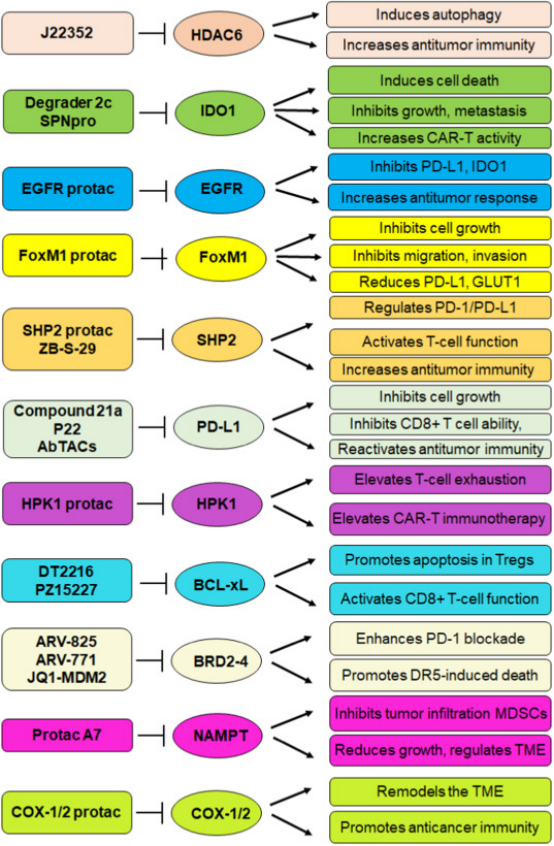
Figure 13. Signaling Pathways by Which PROTACs Enhance the Efficacy of Anticancer Immunotherapy [16]
2. Developing PROTAC Delivery System
Drug delivery platforms enhance the clinical viability of PROTACs by addressing their limitations and maximizing their strengths. The classification of PROTAC delivery systems includes (a) PROTAC prodrug delivery systems, which deliver inactive precursor drugs that become active in the target environment; (b) Nanoplatforms for targeted PROTAC delivery, focusing on precision targeting at the molecular level; (c) Protein-targeted PROTAC delivery systems; (d) Nucleic acid-targeted delivery systems, each designed to optimize delivery and efficacy of PROTACs in various therapeutic contexts.
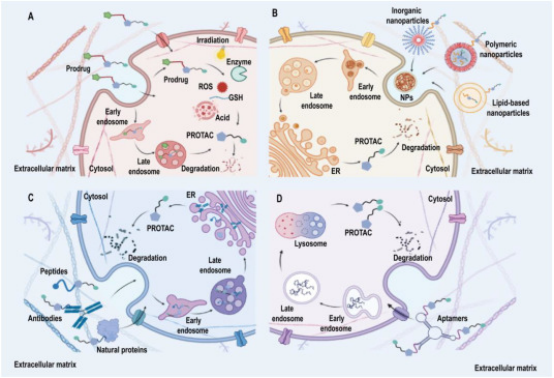
Figure 14. Enhancing Tumor Immunotherapy with PROTACs [6]
3. Computer-Aided PROTAC Screening
Computational techniques are increasingly used to screen for effective PROTACs. Initially, suitable E3 ligases and the molecular fragments that bind to the POI and the ligase are identified. PROTAC molecules are then synthesized by combining these fragments with appropriate linkers, sourced either from existing libraries or designed using generative algorithms. These algorithms are tailored to create compounds with specific attributes, such as desired geometric properties, solubility, permeability, and other drug design-related features like toxicity and synthesizability. Advanced algorithms then predict and rank the stability of the resultant ternary complexes, facilitating the selection of the most promising PROTAC candidates from the library.
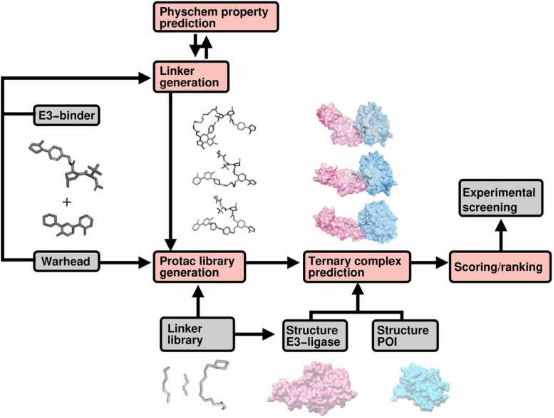
Figure 15. Workflow of Computer-Aided PROTAC Screening [17]
FAQ
Q1: What are the various types of PROTACs?
Numerous specific PROTACs have been developed, including BCL-XL PROTACs, aptamer-PROTAC conjugates (APCs), hypoxia-activated PROTACs, folate-caged PROTACs, antibody-PROTAC conjugates (Ab-PROTACs), and photochemically controllable PROTACs (PHOTACs). These PROTACs are activated by various factors such as enzymatic action and ultraviolet light exposure, enhancing their molecular activity under specific conditions.
References
[1] Shen F, Dassama LMK. Opportunities and challenges of protein-based targeted protein degradation. Chem Sci. 2023 Jul 3;14(32):8433-8447. doi: 10.1039/d3sc02361c. PMID: 37592990; PMCID: PMC10430753.
[2] Amirian R, Azadi Badrbani M, Izadi Z, Samadian H, Bahrami G, Sarvari S, Abdolmaleki S, Nabavi SM, Derakhshankhah H, Jaymand M. Targeted protein modification as a paradigm shift in drug discovery. Eur J Med Chem. 2023 Nov 15;260:115765. doi: 10.1016/j.ejmech.2023.115765. Epub 2023 Aug 29. PMID: 37659194.
[3] Fang Y, Wang S, Han S, Zhao Y, Yu C, Liu H, Li N. Targeted protein degrader development for cancer: advances, challenges, and opportunities. Trends Pharmacol Sci. 2023 May;44(5):303-317. doi: 10.1016/j.tips.2023.03.003. PMID: 37059054.
[4] Sincere NI, Anand K, Ashique S, Yang J, You C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules. 2023 May 10;28(10):4014. doi: 10.3390/molecules28104014. PMID: 37241755; PMCID: PMC10224132.
[5] Sun, X., Gao, H., Yang, Y. et al. PROTACs: great opportunities for academia and industry. Sig Transduct Target Ther 4, 64 (2019). https://doi.org/10.1038/s41392-019-0101-6.
[6] Li Q, Zhou L, Qin S, Huang Z, Li B, Liu R, Yang M, Nice EC, Zhu H, Huang C. Proteolysis-targeting chimeras in biotherapeutics: Current trends and future applications. Eur J Med Chem. 2023 Sep 5;257:115447. doi: 10.1016/j.ejmech.2023.115447. Epub 2023 May 10. PMID: 37229829.
[7] He, M., Cao, C., Ni, Z. et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Sig Transduct Target Ther 7, 181 (2022). https://doi.org/10.1038/s41392-022-00999-9.
[8] Choudhary D, Kaur A, Singh P, Chaudhary G, Kaur R, Bayan MF, Chandrasekaran B, Marji SM, Ayman R. Target protein degradation by protacs: A budding cancer treatment strategy. Pharmacol Ther. 2023 Sep 9;250:108525. doi: 10.1016/j.pharmthera.2023.108525. Epub ahead of print. PMID: 37696366.
[9] Amirian R, Azadi Badrbani M, Izadi Z, Samadian H, Bahrami G, Sarvari S, Abdolmaleki S, Nabavi SM, Derakhshankhah H, Jaymand M. Targeted protein modification as a paradigm shift in drug discovery. Eur J Med Chem. 2023 Nov 15;260:115765. doi: 10.1016/j.ejmech.2023.115765. Epub 2023 Aug 29. PMID: 37659194.
[10] Mi D, Li Y, Gu H, Li Y, Chen Y. Current advances of small molecule E3 ligands for proteolysis-targeting chimeras design. Eur J Med Chem. 2023 Aug 5;256:115444. doi: 10.1016/j.ejmech.2023.115444. Epub 2023 May 8. PMID: 37178483.
[11] Bemis TA, La Clair JJ, Burkart MD. Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J Med Chem. 2021 Jun 24;64(12):8042-8052. doi: 10.1021/acs.jmedchem.1c00482. Epub 2021 Jun 9. PMID: 34106704.
[12] Zografou-Barredo NA, Hallatt AJ, Goujon-Ricci J, Cano C. A beginner's guide to current synthetic linker strategies towards VHL-recruiting PROTACs. Bioorg Med Chem. 2023 Jun 6;88-89:117334. doi: 10.1016/j.bmc.2023.117334. Epub 2023 May 18. PMID: 37224698.
[13] Zhou L, Yu B, Gao M, Chen R, Li Z, Gu Y, Bian J, Ma Y. DNA framework-engineered chimeras platform enables selectively targeted protein degradation. Nat Commun. 2023 Jul 27;14(1):4510. doi: 10.1038/s41467-023-40244-7. PMID: 37495569; PMCID: PMC10372072.
[14] Pu C, Liu Y, Deng R, Xu Q, Wang S, Zhang H, Luo D, Ma X, Tong Y, Li R. Development of PROTAC degrader probe of CDK4/6 based on DCAF16. Bioorg Chem. 2023 Sep;138:106637. doi: 10.1016/j.bioorg.2023.106637. Epub 2023 May 27. PMID: 37276679.
[15] Zhu L, Hu S, Yan X, Zeng Q, Zhang B, Jiang L, Yao SQ, Ge J. Ugi reaction-assisted assembly of covalent PROTACs against glutathione peroxidase 4. Bioorg Chem. 2023 May;134:106461. doi: 10.1016/j.bioorg.2023.106461. Epub 2023 Mar 11. PMID: 36924654.
[16] Li S, Chen T, Liu J, Zhang H, Li J, Wang Z, Shang G. PROTACs: Novel tools for improving immunotherapy in cancer. Cancer Lett. 2023 Apr 28;560:216128. doi: 10.1016/j.canlet.2023.216128. Epub 2023 Mar 16. PMID: 36933781.
[17] Tunjic TM, Weber N, Brunsteiner M. Computer aided drug design in the development of proteolysis targeting chimeras. Comput Struct Biotechnol J. 2023 Feb 24;21:2058-2067. doi: 10.1016/j.csbj.2023.02.042. PMID: 36968015; PMCID: PMC10030821.
How to order?

