





N-Terminal Sequence Analysis Service
- Enables acquisition of extensive N-terminal sequence information.
- Can analyze proteins with N-terminal blocking and modifications.
- Cannot determine equimolar amino acids.
- Peptide sequence coverage is seldom 100%.
- Can accurately analyze N-terminal amino acid sequences, especially in differentiating isoleucine from aspartic acid, and glutamine from lysine.
- Directly analyzes amino acid information without the need for any protein database, enabling the identification of unknown proteins not registered in databases.
- Peptides with chemically modified or blocked N-terminal are not amenable to analysis.
- Low throughput.
The N-terminal sequence significantly influences a protein's half-life and its subcellular localization. This region is pivotal for the structural and functional integrity of proteins and peptides. Analyzing the N-terminal sequence enables identification of the protein's initiation point, facilitating insights into its higher-order structure and biological functions. For purified protein products, antibodies, and protein vaccines, the accuracy, stability, and consistency of the N-terminal sequence are critical. Predominant methods for determining the N-terminal of proteins or peptides encompass the Sanger method, Enzymatic digestion, Edman degradation, 2,4-dinitrofluorobenzene (DNFB) method, Dansyl(DNS) method, and Mass spectrometry. Edman degradation and Mass spectrometry are the principal techniques for N-terminal sequence analysis.
MtoZ Biolabs employs the Shimadzu Edman sequencing system for N-terminal sequencing of purified protein products, antibodies, and protein vaccines, catering to researchers and scientific clientele. Furthermore, Mtoz Biolabs has developed a platform utilizing advanced LC-MS/MS technology for N-terminal sequencing, adept at detecting blocked and modified protein termini. This methodology enhances the Edman degradation process, facilitating comprehensive N-terminal sequencing services.
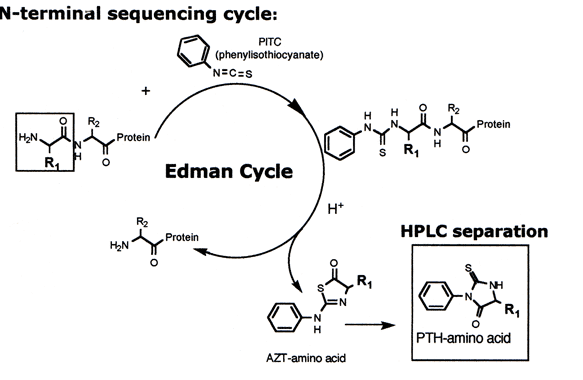
1. Edman Degradation
Edman sequencing, a cyclic process conducted under mildly alkaline conditions, involves the reaction of a polypeptide/protein with phenyli sothiocyanate (PITC). Following acidification, the N-terminal amino acid residue is released as a phenylthiohydantoin (PTH) derivative, subsequently separated and identified. This technique permits sequential identification of amino acids from a peptide or protein's N-terminal.
2. Mass Spectrometry
N-terminal sequencing via mass spectrometry entails employing two proteases with complementary cleavage specificity to digest the target protein, producing two N-terminal peptides of varying lengths. These peptides, suitable for ionization, undergo mass spectrometric analysis. Comparison with the theoretical amino acid sequence facilitates deduction of the N-terminal peptides' sequences, with the enzymatic digestions providing mutual validation and confirming the protein's N-terminal sequence.
Analysis Workflow
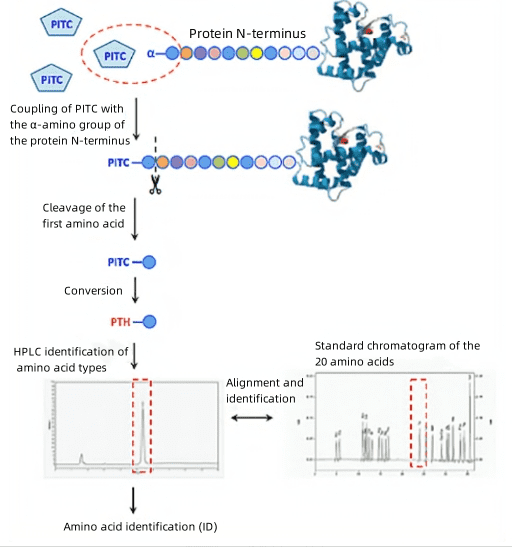
1. Protein N-terminal Sequencing via Edman Degradation
(1) Separate protein samples via SDS-PAGE to verify sequencing purity requirements.
(2) Transfer proteins from SDS-PAGE gel to a PVDF membrane, visualize through staining, and excise.
(3) Conduct analysis using an Edman sequencer on the PVDF membrane samples.
(4) For higher purity samples, dot blotting can be performed directly on the membrane protein samples, which can then be analyzed using an Edman sequencer.
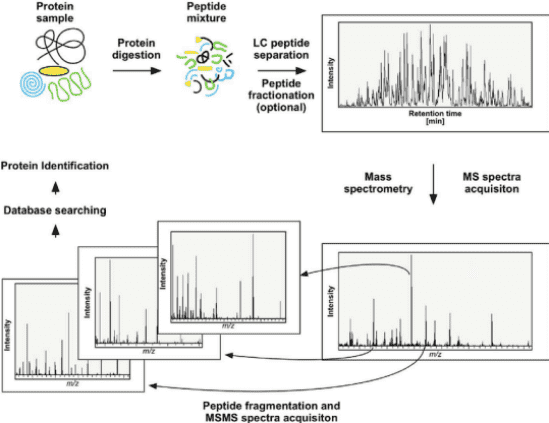
2. Protein N-terminal Sequencing via LC-MS/MS
(1) Digest the protein samples into a mixture of peptides through enzymatic digestion.
(2) Desalt the peptide mixture and perform tandem mass spectrometry usingQ-Exactive series mass spectrometer (Thermo Fisher Scientific).
(3) Match the spectra against the theoretical amino acid sequences of the proteins to determine the complete N-terminal amino acid sequence.
Service Advantages
1. Offers a comprehensive methodology, encompassing a wide array of protein sample N-terminal sequence analyses.
2. Edman degradation enables precise N-terminal amino acid sequence analysis without relying on protein databases, distinctly identifying isoleucine from leucine and glutamine from lysine.
3. Edman degradation can produces chromatograms for each amino acid at the N-terminal.
4. Utilizes software in mass spectrometry to align mass spectral data with theoretical sequences, accurately locating the N-terminal in protein samples and providing detailed secondary mass spectra for the N-terminal.
Sample Results
1. Edman Degradation
Initial calibration for the 19 PTH amino acids involves a calibration test using a mixed standard of all 19 PTH amino acids. The calibration test chromatogram is displayed as follows:
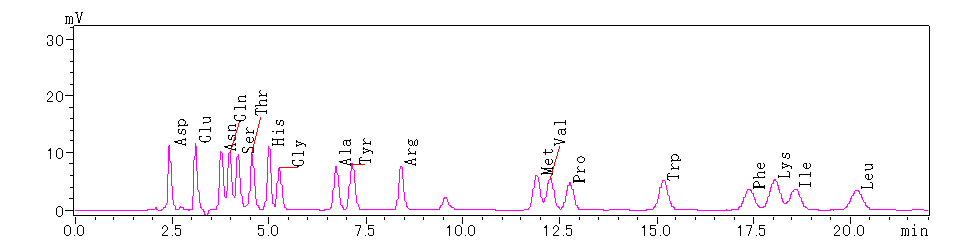
Figure 1. Calibration Test Chromatogram for Standard Mixture
Following the completion of the calibration test for the PTH amino acid mixed standard, the N-terminal sequence of the test sample is analyzed. The chromatograms for the test sample are as follows:

Figure 2. N-terminal 1st Amino Acid
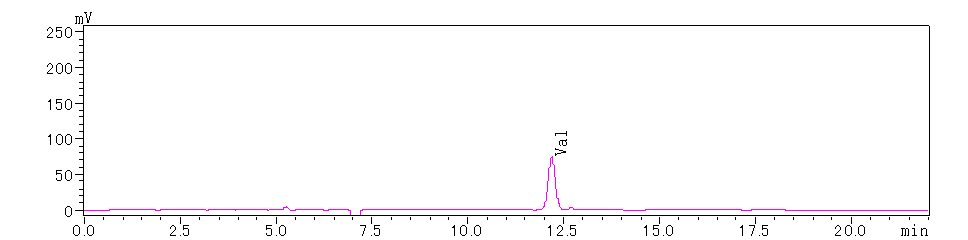
Figure 3. N-terminal 2nd Amino Acid

Figure 4. N-terminal 3rd Amino Acid
Based on the above chromatograms, the N-terminal sequence of the protein is determined to be Tyr-Val-Trp.
2. Mass Spectrometry
Post-trypsin digestion, LC-MS/MS analysis of the sample yields mass spectrometry data. Correlating this data with a theoretical database produces an MS2 spectrum aligning with the theoretical N-terminal sequence (HYAHVDCPGHADYVK), as shown below:
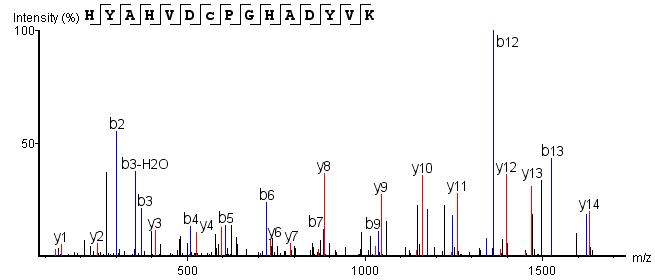
Figure 5. N-terminal Sequence MS2 Spectrum
From this analysis, the N-terminal starting amino acid of the monoclonal antibody drug is identified as histidine (H).
Sample Submission Requirements
1. Edman Degradation
(1) The required sample quantity ranges from 20 µg to 50 µg.
(2) The purity of the sample must exceed 90%.
(3) Do not use surfactants in the buffer, and minimize the salt concentration of the solution as much as possible.
(4) Compounds such as Tris, glycine, guanidine, glycerol, sucrose, ethanolamine, SDS, Triton X-100, Tween, and other detergents, along with ammonium sulfate and other ammonium salts, can adversely affect the subsequent amino acid identification. Their use should be avoided during sample preparation.
(5) Throughout the sample preparation process, it is imperative to wear masks and hair covers to prevent contamination from keratin.
2. Mass Spectrometry
(1) Both in-gel samples and solution samples are suitable for the aforementioned N-terminal sequencing.
(2) The target protein sample required is between 5 µg and 10 µg.
(3) The higher the purity of the sample, the better.
Services at MtoZ Biolabs
1. Project Report
Experimental Procedures, Relevant Mass Spectrometry Parameters, Detailed Information on the N-terminal Sequence of Proteins, and Mass Spectrometry Images.
2. Raw Data
FAQ
Q1: What are the advantages and disadvantages of Edman degradation and mass spectrometry in detecting the N-terminal of proteins?
(1) Mass Spectrometry
① Advantages
② Disadvantages
(2) Edman Degradation
① Advantages
② Disadvantages
Q2: How to determine the N-terminal position of proteins with N-terminal modifications?
During data analysis, analyze the N-terminal PTMs simultaneously to identify the modified N-terminal positions.
How to order?

