





Antibody De Novo Sequencing Service for R&D | Mass Spec
Antibodies bind to various antigens with high affinity and specificity, playing a crucial role in adaptive immune response to infections and can also mediate autoimmune diseases by targeting self-antigens. Antibodies facilitate immunity by inhibiting critical stages of a pathogen's replication cycle, such as receptor binding and cellular entry, activating the complement system, or initiating specific cell-mediated immune response, such as antibody-dependent cellular cytotoxicity. Antibodies can persist in circulation for months and swiftly regenerate upon subsequent antigen exposure through memory B cell responses. These properties establish antibodies as vital serological markers for pathogen exposure and vaccine efficacy, therapeutic leads in cancer and infectious disease treatment, and invaluable tools for specific labeling and molecular target detection. Identifying therapeutic and intrinsic soluble antibodies primarily involves determining their amino acid sequences, typically achieved through sequencing the B cell receptor complex at the nucleic acid level, with mass spectrometry (MS) serving as another method for De Novo protein-level sequence information acquisition.
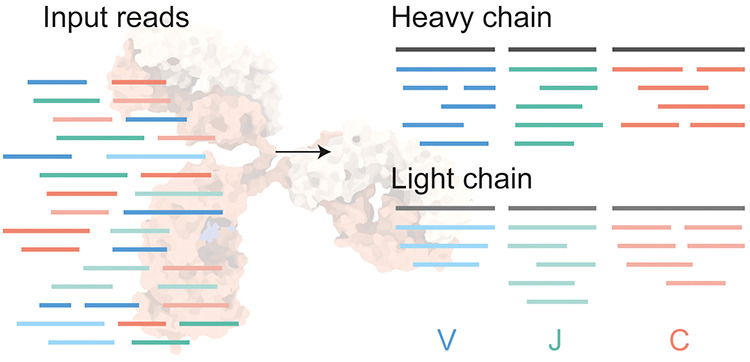
Figure 1. De Novo Antibody Sequencing[4]
1. Proteomic Methods for De Novo Sequencing
Proteomics involves the extensive study of proteins, developing numerous peptide- and protein-focused MS-based techniques, including those for de novo sequence analysis of antibody. The most common MS-based protein analysis technique, bottom-up MS, involves digesting proteins with proteases, separating the resulting peptides via liquid chromatography (LC), and recording their masses (MS1 and MS2 for peptide and fragment ions, respectively). The predictability of peptide and fragment ion generation allows for identification by matching recorded spectra with simulated spectra from protein or DNA databases. In antibody sequencing, customized databases are crucial for accurate identification. Even without such databases, the digestion-based strategy is extensively utilized for de novo sequencing of individual spectra, compiling the reads into full-length sequences. Intact mass analysis, comparing various masses, yields insights into differences caused by known mutations, post-translational modifications, or signal peptide. Analyzing antibodies in their native or denatured states facilitates understanding of their structural complexity or changes in abundance of specific clones. Knowledge of precursor mass is instrumental in accurately predicting light and heavy chain sequences in de novo sequencing. Denatured and native antibodies can also be sequenced through fragmentation, known as top-down MS. This method deals with larger, more highly charged molecular species, resulting in complex, such intact-protein fragmentation spectra are more complex and harder to interpret than peptide spectra. Middle-down MS, which involves cleaving antibodies at the heavy chain's hinge region before analysis, simplifies these challenges, enabling effective sequence determination and verification.
In recent years, significant advancements in sample preparation, MS and LC instrumentation, and data analysis have been achieved in MS-based proteomics. Using all these advances, antibody sequencing at the protein level by MS has come within reach. There are currently three main strategies for antibody sequencing: (1) The first strategy uses bottom-up MS for the full-length sequencing of recombinant antibodies, typically requiring large quantities of highly purified monoclonal antibodies (mAbs). Digesting mAbs with one or several proteases yields hundreds of peptides, which provide multiple overlapping short sequence reads. These peptides are analyzed by LC-MS, then processed and assembled into the full-length mAb sequence using various de novo sequencing software solutions. (2) The second strategy employs a hybrid approach that integrates MS-based techniques with genomic/transcriptomic analysis (e.g., whole-genome sequencing or BCR sequencing, ideally from the same donor) to study the endogenous antibody repertoire. This involves creating a sequence database through B-cell sequencing and acquiring MS data through bottom-up MS. Although this approach relies on identifying antibodies from a pre-existing sequence database, making it not strictly de novo, it serves as a powerful tool for antibody library analysis. (3) The third approach encompasses various MS-based de novo research methods designed to directly determine complete antibody sequences of selected clones from clinical samples. Endogenous antibody sequencing cannot depend solely on top-down MS due to the inherent challenges and complexities of direct sequencing from endogenous fluid antibodies. Newly developed middle-down or top-down proteomic techniques provide clone-specific sequence information, effectively complementing traditional sequencing methods. By integrating various proteomic techniques, it is possible to achieve comprehensive full-length coverage of antibody sequences.
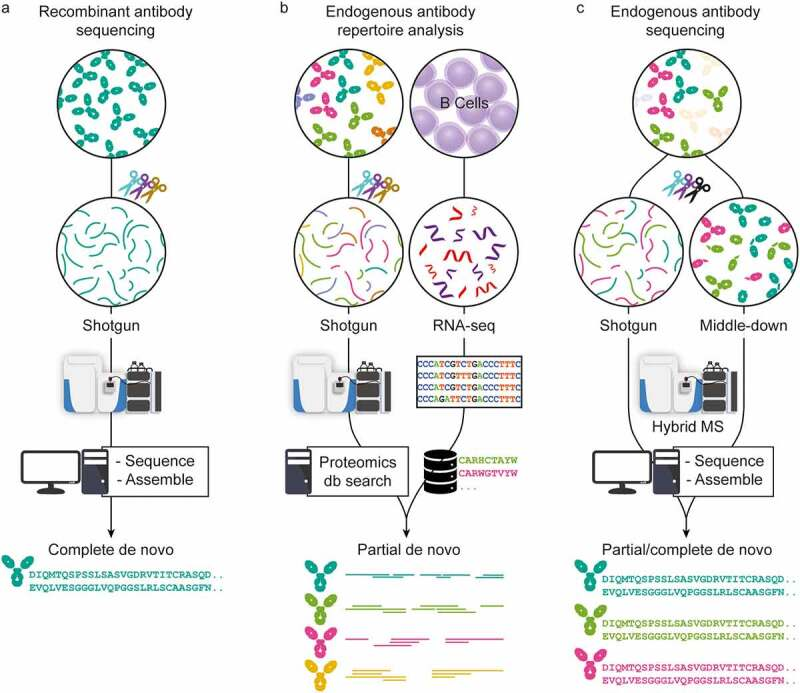
Figure 2. Three Approaches to MS-Based Antibody Sequencing [5]
2. Peptide De Novo Sequencing Algorithms
Due to the large number of spectra generated in a single LC-MS/MS run, algorithms are essential for assigning peptide fragment sequences in MS/MS for automated analysis. The development of these algorithms began in the 1980s. The initial approach was an exhaustive search, which involved listing every possible amino acid combination with masses matching the precursor ion mass and comparing these with the observed spectra. This method incorporated a scoring function to evaluate how well the tentative sequences matched the experimental data, with the sequence scoring the highest selected as the correct sequence.
As collection technologies and instrumentation have evolved, it became feasible to acquire MS/MS spectra of longer peptides at higher throughput, rendering the exhaustive search computationally impractical. Consequently, more sophisticated algorithms for peptide sequencing have emerged, broadly categorized into graph-based, dynamic programming, and machine learning/neural network algorithms.
(1) Graph-based Algorithms: Since the 1990s, algorithms based on graph theory have been developed for sequencing reconstruction. Briefly, the m/z values of spectral peaks are transformed into vertices of a directed acyclic graph (DAG). Vertices are connected if the mass difference between them equals the mass of an amino acid residue. The longest path in this graph corresponds to the optimal peptide sequence. Algorithms such as Sherenga and SeqMS have been enhanced by considering the intensity of peaks. Methods that prioritize the highest intensity peaks identify the m/z values corresponding to the first or second highest intensities to establish clear amino acid labels. Examples of such labeling algorithms include GutenTag and DirecTag. Graph theory is also employed to distinguish between b and y ions, simplifying the spectra and enhancing accuracy by minimizing the selection of unrelated peaks or noise. NovoHCD integrates peptide tags, amino acid combinations (AAC), and a refined graph model with multiple edge types to analyze HCD spectra. Similarly, NovoGMET is designed for ExD spectra analysis. Additionally, for more effective analysis of cyclic peptide spectra, the specialized CycloNovo algorithm uses a de Bruijn graph.
(2) Dynamic Programming Algorithms: Dynamic programming algorithms are selected to decrease the computational time for spectra analysis. PEAKS, a commercial software package, employs dynamic programming for sequencing and introduces a probabilistic scoring scheme for choosing sequences. Pepnovo's scoring method reflects peptide fragmentation rules and tests the likelihood of observed peaks being model-based rather than random. MSNovo also adopts dynamic programming and excels with ion trap data. pNovo is developed for analyzing HCD spectra, taking into account spectral features absent in traditional CID to improve sequence accuracy. Its successor, pNovo+, is designed for simultaneous analysis of HCD and ETD spectra to exploit their complementary strengths. CompNovo is another algorithm capable of analyzing multiple spectral inputs. This algorithm initially preprocesses the spectra, then applies a divide-and-conquer strategy to break down the spectra into smaller segments until they are sufficiently small for mass analysis to generate all possible amino acid compositions for a given mass difference. Uninovo integrates common fragmentation techniques into one software package to enhance sequencing accuracy.
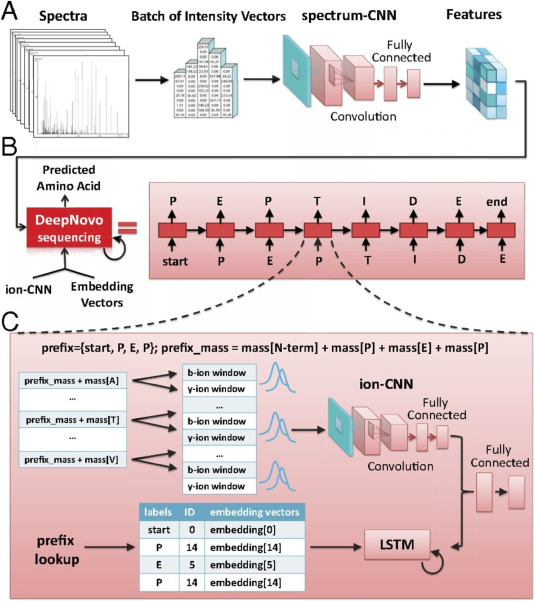
Figure 3. Neural Network-Based DeepNovo Algorithm [6]
(3) Machine Learning and Neural Network Algorithms: Machine learning algorithms are employed for MS/MS spectra analysis. Typically, extensive annotated spectra from a spectral database are used to train these algorithms. The training process involves using regression and statistical methods such as tree-based or support vector models to develop predictive models. These models are then utilized to validate test spectra. NovoHMM, the first algorithm to employ a hidden markov model (HMM)-a frequent method in machine learning-simulates the generation of MS/MS spectra probabilistically and outperforms existing resequencing tools at that time. Novor features a novel scoring system developed from decision trees trained on an existing spectral library through machine learning, incorporating knowledge of trained peptide fragments. Novor sequences faster than some mass spectrometers' acquisition speeds, facilitating real-time de novo sequencing. Neural networks, inspired by our brain's biological neurons, represent a hierarchical structure that better models nonlinear systems. Deep learning, an evolution of neural networks, has been proven in fields like image recognition and autonomous driving. In de novo sequencing, various algorithms leverage neural networks' object recognition capabilities for MS/MS spectra analysis. Recurrent neural networks (RNNs) are applied to predict MS/MS spectra, while convolutional neural networks (CNNs), successful in image recognition, are adapted for spectral analysis.
3. Applications of De Novo Antibody Sequencing in Drug Research and Development
De novo antibody sequencing is increasingly used to identify the sequences and sequence variants of original biologics, aiding in the development of biosimilars. In this context, de novo sequencing is pivotal for the initial identification and validation of the amino acid sequences of original biologics. Moreover, de novo antibody sequencing offers two particularly valuable applications for biotech/biopharmaceutical companies in the discovery phase: 1) isotype selection and 2) bispecific antibody concept verification. The objective of isotype selection is to explore the influence of isotypes on the functionality of alternative antibodies. Through de novo sequencing, teams can sequence commercial antibodies targeting mouse antigens and recombine these to produce chimeric antibodies with various human Fc variants. These antibodies are then tested both in vivo and in vitro in transgenic mice, assessing the isotype's impact on antibody functionality. Alternative methods, such as acquiring original commercial antibody hybridoma cell lines or starting new antibody development programs, would be more costly and time-consuming compared to de novo sequencing, making it an efficient and cost-effective solution.
mAbs are dimers with both arms binding to the same antigen. Bispecific antibodies can bind to more than one antigen, which has sparked significant interest due to their potential for new therapeutic target interactions. Selecting combinations of targets with optimal therapeutic outcomes is a complex challenge. In some instances, bispecific antibodies have proven more effective than combination therapies, though there have been instances of less favorable outcomes. Consequently, meticulous selection and screening of target combinations are imperative. By employing novel antibody sequencing technologies, a range of bispecific concept verification molecules can be created to identify the most effective target combinations. Using antibodies that are commercially available and have been functionally validated, it is possible to generate recombinant bispecific antibodies that concurrently target two antigens. Through subsequent in vivo and in vitro testing in primary mouse models and human trials, the most synergistic target combinations can be identified, facilitating the initiation of therapeutic endeavors and reducing the project's risk. Without de novo sequencing, such an approach would be impractical.
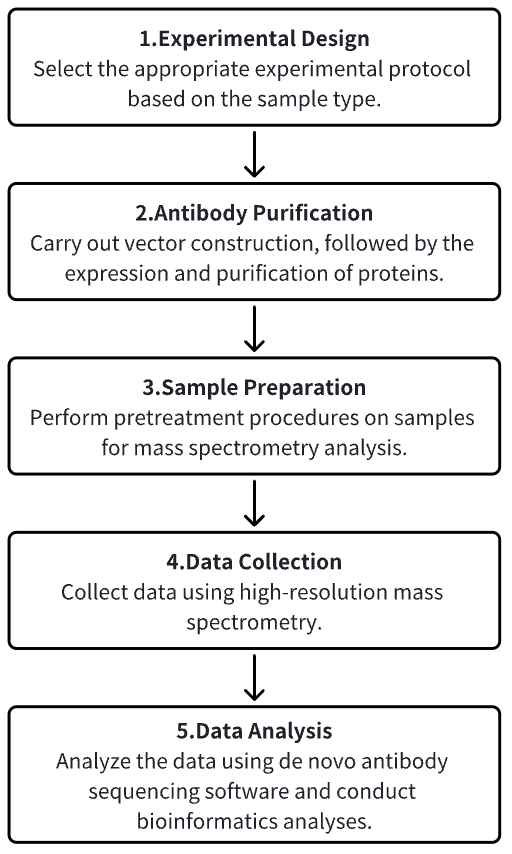
Analysis Workflow
1. Establishment of the Experimental Protocol Based on Experimental Needs
2. Antibody Purification
3. Sample Preparation for Mass Spectrometry Analysis
4. Data Collection Using High-Resolution Mass Spectrometry
5. Data Retrieval and Analysis
Service Advantages
1. Integrated Services for Antibody Expression, Purification, and Preparation of Mass Spectrometry Samples
2. Highly Reliable and Precise Mass Spectrometry Analysis
3. Extensive Bioinformatics Analysis
Example Results
1. Mass Spectrometry-Based De Novo Sequencing Employing Multiple Proteases and a Dual Fragmentation Scheme for Monoclonal Antibodies
Understanding the sequence of antibodies is essential for grasping the structural basis of antigen binding, a key aspect of therapeutic and research applications of antibodies. A method allowed for direct de novo sequencing of monoclonal IgG from purified antibody products. This approach utilized several complementary proteases—trypsin, chymotrypsin, lysN, lysC, gluC, aspN, aLP, thermolysin, and elastase-to generate peptides apt for LC-MS/MS de novo sequencing in a bottom-up fashion. Furthermore, peptide precursors undergo stepped high energy collision dissociation (stepped HCD) and electron-transfer high-energy collision dissociation (EThcD) in a dual fragmentation scheme. This technique achieved full sequence coverage of the mAb Herceptin, with a 99% accuracy in the variable regions. Applied to the widely-used anti-FLAG-M2 mouse mAb, this method confirmed its efficacy by sequencing and validating its functionality through the reconstruction of the Fab's high-resolution crystal structure and its binding with FLAG-tagged target proteins in Western blot analysis. Thus, this method ensured robust and reliable sequencing of mAbs.
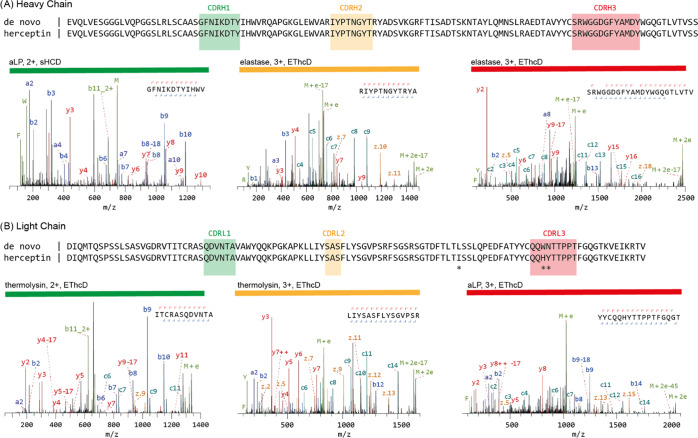
Figure 4. De Novo Sequencing of Monoclonal Antibody Herceptin via MS [7]
2. Template-Based Assembly of Proteomic Short Reads for De Novo Antibody Sequencing and Sequence Analysis
Antibodies target various antigen molecules through the diversity generated by somatic recombination and hypermutation. To fully understand the role of antibodies in health and disease, specialized de novo sequencing methods are essential. Although next-generation cDNA sequencing has established a basis for exploring the antibody repertoire, it primarily targets the B cells that produce the antibodies, not the secreted polypeptide products. MS-based methods can directly acquire sequence information from these secreted peptides, potentially closing the gap between antibody repertoire profiling and bulk serological assays. To tackle the challenges of MS-based antibody sequencing, research has introduced a swift, straightforward software tool named Stitch, tailored for mapping proteomic short reads onto user-defined templates. Its functions are ideal for sequencing both mAbs and polyclonal antibody repertoires, as well as for repertoire analysis. Stitch has proven effective by fully reconstructing two mAbs sequences with greater than 98% accuracy (including I/L assignment), sequencing Fabs from patient serum against a high background of homologous antibody sequences, and analyzing light chains in the urine of multiple myeloma patients and IgG repertoire in sera from patients hospitalized with COVID-19. Stitch's capabilities enable comprehensive analysis of antibody sequences, representing a significant advancement in polyclonal antibody and repertoire analysis.
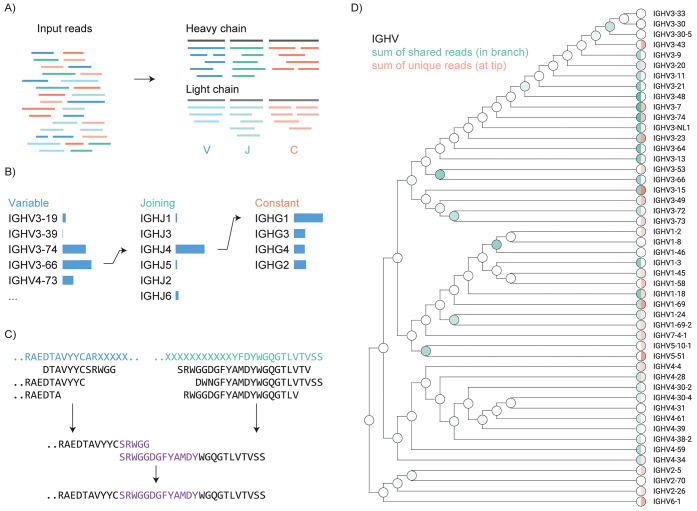
Figure 5. Schematic Overview of the Stitch Software [4]
3. Direct Determination of Antibody Chain Pairing Using Electron Capture Dissociation and Ultraviolet Photodissociation through Top-Down and Middle-Down Mass Spectrometry
A significant challenge in the development and discovery of mAb therapeutics is identifying the pairing between heavy and light chains. Technological advances in MS and MS/MS techniques have significantly enhanced the capability to analyze large, intact proteins, thus enabling more detailed and accurate characterization of protein conformations. Directly detecting intact antibodies or fragments such as F(ab′)2 and Fab fragments can substantially streamline therapeutic mAb discovery processes. Recent studies have demonstrated the efficacy of electron capture dissociation (ECD) and 157 nm ultraviolet photodissociation (UVPD) in effectively cleaving disulfide bonds that link mAb heavy and light chains. By analyzing the total mass of mAbs, Fabs, or F(ab′)2, along with the mass of complete light chains and fragment of domain (Fd), and covering CDR3 sequences, it was possible to determine the chain pairing with a single experimental setup. These findings highlighted the role of top-down and middle-down MS in significantly simplifying the discovery of therapeutic antibodies.
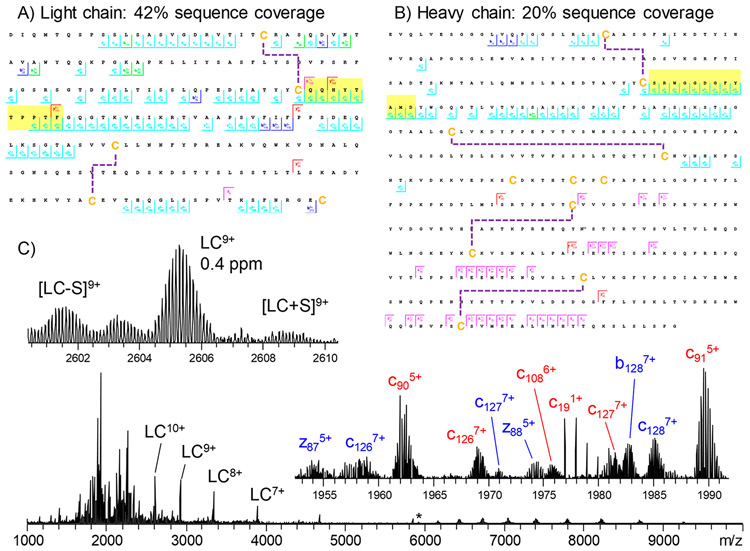
Figure 6. Sub-Ion Map of Rituximab Light Chain and a Glycosylated Heavy Chain with G0F, Showing the Sequence Coverage Achieved by EChcD Collision Energy[8]
Sample Submission Requirements
1. Protein Purity > 90%
2. Minimize Impurity Contamination
Services at MtoZ Biolabs
1. Complete Experimental Steps
2. Relevant Instrument Parameters
3. Original Experimental Data
4. Data Analysis Report
Applications
1. Sequencing of Human Plasma Antibody Libraries
Billions of IgG1 variants can be produced in humans through recombination and hypermutation. However, the diversity of circulating IgG1 clones in human plasma has largely remained uncharacterized. Several MS-based methods have been employed to reveal that the IgG1 gene library in the plasma of healthy donors and sepsis patients is predominantly composed of a limited number of clones. The studies demonstrated that each donor displayed a unique serological IgG1 rearrangement which is stable over time, yet capable of rapidly adapting to physiological changes.
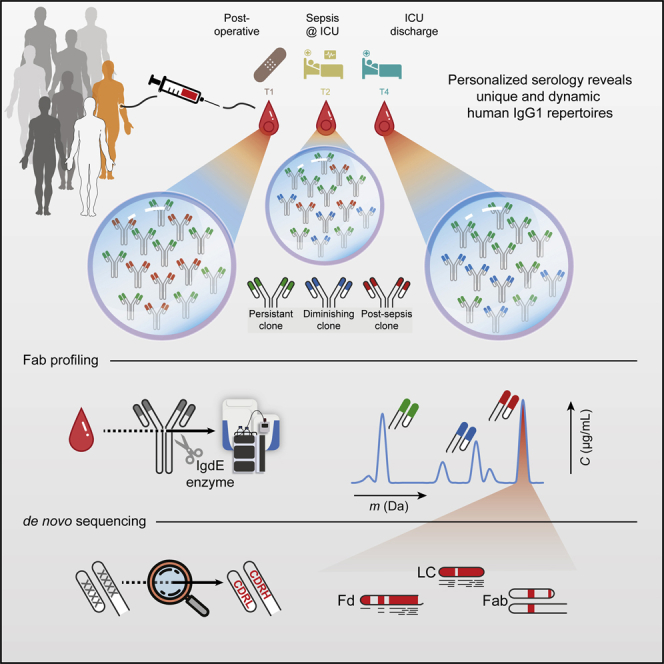
Figure 7. Human Plasma Antibody Library [9]
FAQ
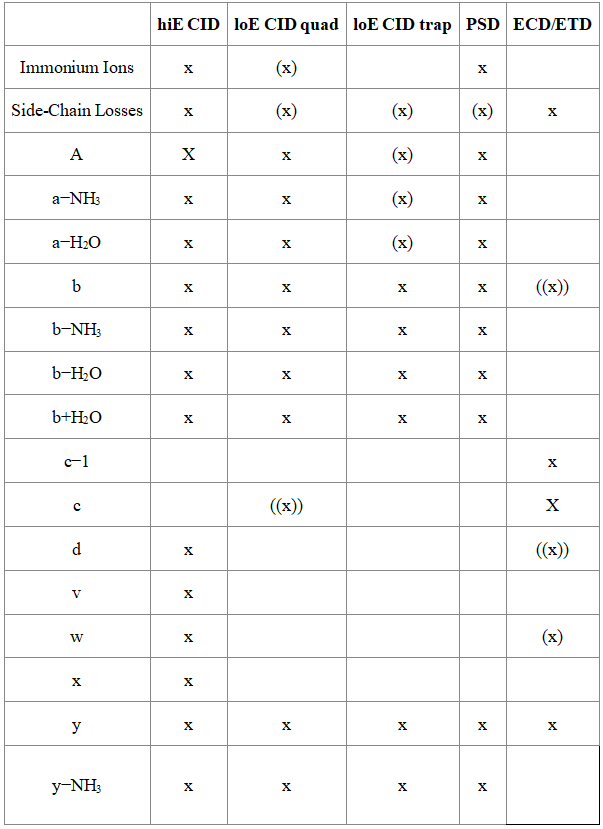
Q1: What ion types are produced by various fragmentation methods?
Various MS/MS experiments have observed the following types of fragment ions:
References
[1] Snapkov I, Chernigovskaya M, Sinitcyn P, Lê Quý K, Nyman TA, Greiff V. Progress and challenges in mass spectrometry-based analysis of antibody repertoires. Trends Biotechnol. 2022 Apr;40(4):463-481. doi: 10.1016/j.tibtech.2021.08.006. Epub 2021 Sep 14. PMID: 34535228.
[2] de Brito PM, Saruga A, Cardoso M, Goncalves J. Methods and cell-based strategies to produce antibody libraries: current state. Appl Microbiol Biotechnol. 2021 Oct;105(19):7215-7224. doi: 10.1007/s00253-021-11570-x. Epub 2021 Sep 15. PMID: 34524471.
[3] Alejandra WP, Miriam Irene JP, Fabio Antonio GS, Patricia RR, Elizabeth TA, Aleman-Aguilar JP, Rebeca GV. Production of monoclonal antibodies for therapeutic purposes: A review. Int Immunopharmacol. 2023 Jul;120:110376. doi: 10.1016/j.intimp.2023.110376. Epub 2023 May 25. PMID: 37244118.
[4] Schulte D, Peng W, Snijder J. Template-Based Assembly of Proteomic Short Reads For De Novo Antibody Sequencing and Repertoire Profiling. Anal Chem. 2022 Jul 26;94(29):10391-10399. doi: 10.1021/acs.analchem.2c01300. Epub 2022 Jul 14. PMID: 35834437; PMCID: PMC9330293.
[5] de Graaf SC, Hoek M, Tamara S, Heck AJR. A perspective toward mass spectrometry-based de novo sequencing of endogenous antibodies. MAbs. 2022 Jan-Dec;14(1):2079449. doi: 10.1080/19420862.2022.2079449. PMID: 35699511; PMCID: PMC9225641.
[6] Ng CCA, Zhou Y, Yao ZP. Algorithms for de-novo sequencing of peptides by tandem mass spectrometry: A review. Anal Chim Acta. 2023 Aug 8;1268:341330. doi: 10.1016/j.aca.2023.341330. Epub 2023 May 8. PMID: 37268337.
[7] Peng W, Pronker MF, Snijder J. Mass Spectrometry-Based De Novo Sequencing of Monoclonal Antibodies Using Multiple Proteases and a Dual Fragmentation Scheme. J Proteome Res. 2021 Jul 2;20(7):3559-3566. doi: 10.1021/acs.jproteome.1c00169. Epub 2021 Jun 14. PMID: 34121409; PMCID: PMC8256418.
[8] Shaw JB, Liu W, Vasil Ev YV, Bracken CC, Malhan N, Guthals A, Beckman JS, Voinov VG. Direct Determination of Antibody Chain Pairing by Top-down and Middle-down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation. Anal Chem. 2020 Jan 7;92(1):766-773. doi: 10.1021/acs.analchem.9b03129. Epub 2019 Dec 12. PMID: 31769659; PMCID: PMC7819135.
[9] Bondt A, Hoek M, Tamara S, de Graaf B, Peng W, Schulte D, van Rijswijck DMH, den Boer MA, Greisch JF, Varkila MRJ, Snijder J, Cremer OL, Bonten MJM, Heck AJR. Human plasma IgG1 repertoires are simple, unique, and dynamic. Cell Syst. 2021 Dec 15;12(12):1131-1143.e5. doi: 10.1016/j.cels.2021.08.008. Epub 2021 Sep 17. PMID: 34613904; PMCID: PMC8691384.
[10] Medzihradszky KF, Chalkley RJ. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom Rev. 2015 Jan-Feb;34(1):43-63. doi: 10.1002/mas.21406. PMID: 25667941; PMCID: PMC4367481.
How to order?

