





Tumor-Associated Antigens Identification Service
To accelerate the development of cancer immunotherapy, it is essential to systematically identify tumor antigens that are truly tumor-specific, patient-shared, stably expressed (i. e., cloned) and immunogenic in all malignant cells. Immunopeptidomics is a large-scale and in-depth approach capable of identifying and quantifying the presented human leukocyte antigen (HLA) immunopeptidome. However, it requires the use of advanced analytical and computational proteomics pipelines to accurately identify, validate, and screen both typical and atypical antigens for preclinical test and clinical implementation.
The distinction between tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) lies in that the former is exclusively present on tumor cells, while the latter can be found in both normal and tumor cells. Currently, in addition to focusing on classical types of TSAs and TAAs, researchers are shifting attention to cancer antigens previously overlooked, such as non-canonical proteins and bacterial proteins. The types of tumor antigens recognized by T cells can be classified into five categories: (1) Overexpressed tumor and tissue-specific antigens. They exist widely in tumor cells. However, they are also expressed in normal cells. Hence these antigens have low tumor specificity and are hindered by central tolerance. (2) Cancer germline antigens (CGAs). They are expressed only in the germline and re-expressed in tumor cells with moderate tumor specificity and central tolerance. (3) Viral and bacterial antigens. They originate from previous carcinogenic pathogen infections and have no expression in healthy tissues. They are characterized by high tumor specificity and a lack of central tolerance. (4) Neoantigens caused by mutations, which include variants, insertion deletions or fusion of single nucleotide. They originate from tumorigenesis and are present only in cancer cells with high tumor specificity and no central tolerance. (5) Neoantigens derived from aberrant translation or transcription. They are the result of dysfunctions in the cellular transcription and translation mechanisms in cancer and are not encoded by the genome. As a new category of TSAs, their prevalence and tumor specificity largely remain to be explored.
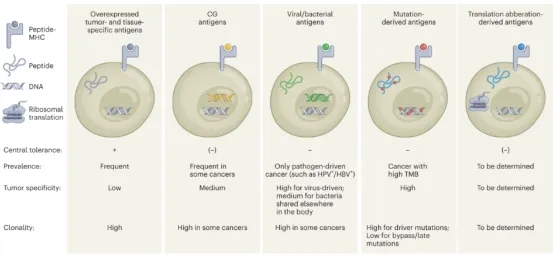
Figure 1. Types of Tumor Antigens Recognized By T Cells [1]
Both the conventional proteasome and the immunoproteasome are involved in the formation of antigenic peptides. The resulting peptides, ranging in size from 9 to 16 amino acid residues, are transported into the lumen of the endoplasmic reticulum by transport proteins associated with the antigen processing (TAP) complex. Newly synthesized MHC class I (MHC I) molecules, together with β2-microglobulin (B2M), tapasin, ERp57 and calreticulin, form a peptide-loading complex (PLC) that facilitates the fold of MHC I. Antigenic peptides undergo additional trimming by the endoplasmic reticulum aminopeptidases 1 (ERAP1) and ERAP2, then are loaded onto the peptide-binding groove of the MHC I. Its structure can be optimized via TAP binding protein-related protein (TAPBPR)-mediated optimization. The stable peptide-MHC I is dissociated from PLC and transported to the cell surface and vesicles through the Golgi apparatus, which is stimulated by IFN γ. Once presented on the surface of tumor cells, the peptides bound can be recognized by the T-cell receptors (TCR) of CD8 T cells to trigger an immune response.
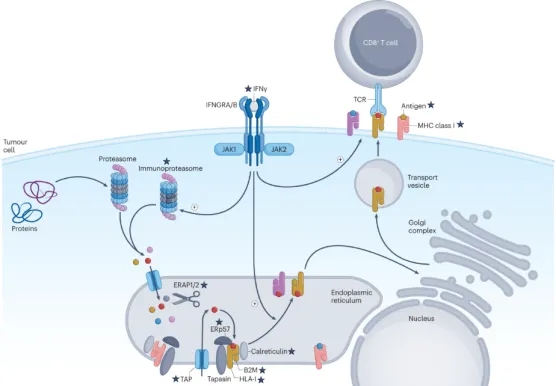
Figure 2. HLA-I Processing and Presentation Pathway [2]
These non-canonical antigens may be the result of alterations in the levels of the genome, epigenome, transcriptome, proteome and antigen processing. For instance, post-transcriptional events such as alternative splicing, intron retention, non-canonical translation initiation, and codon readthrough contribute to the diversity of these antigens. Furthermore, long non-coding RNAs (lncRNAs) and pseudogenes can produce peptides, some of which can stimulate T-cell responses in the reports. Somatic mutations underlying the foreignness of cancer cells may result in alterations to the sequence of protein depending on the type of mutations. A significant area of future development is the utilization of the various categories described above, as well as other categories (such as non-coding regions of the genome), to expand the discovery space for predicting new epitopes.
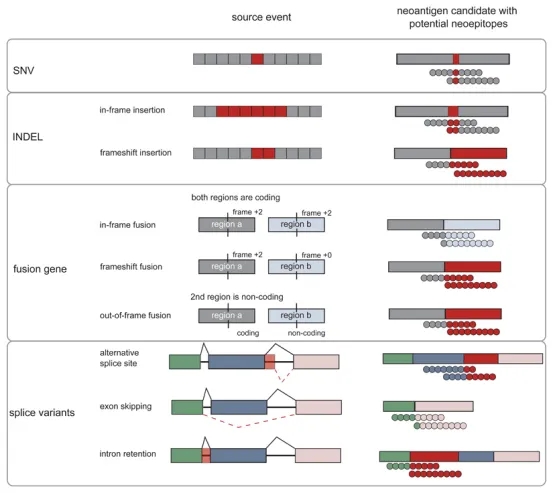
Figure 3. Mutants and Their Derived Tumor Neoantigens and Neoepitopes [3]
After identifying TAAs, clinical studies can be conducted on either universal or personalized peptide vaccines. Following vaccination, both new antigen-specific CD4 T cells and CD8 T cells can be induced de novo or by boosting existing new antigen-specific T-cell responses. These T cells proliferate and kill tumor cells expressing the new antigens. As tumor cells are eliminated, the release of tumor antigens may contribute to epitope spreading, thereby increasing the breadth of tumor-specific T-cell responses. After the eradication of the tumor, the responsive T cell diminishes and persists as part of the memory T-cell repertoire. Vaccine-induced new antigen-specific T cells have the potential to generate long-lived central memory T cells and effector memory T cells.
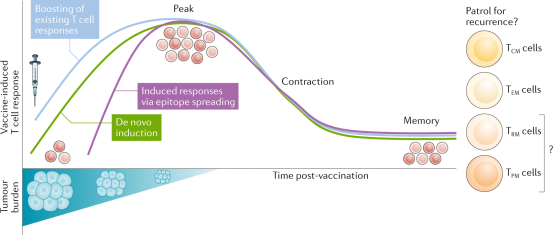
Figure 4. Personalized Vaccines Based on New Antigens Can Induce A Durable Tumor-Specific Memory T-Cell Population [4]
Alterations in antigen processing and presentation caused by germline and somatic changes are crucial for therapeutic responses and patient outcomes following treatment with immune checkpoint inhibitors (ICIs). Given the inter-tumoral heterogeneity among patients, a range of predictive biomarkers, including PD-1 and PD-L1 expression, tumor mutational burden (TMB), MHC class I (MHC I) expression, HLA evolutionary divergence (HED), loss of heterozygosity (LOH) in HLA class I (HLA-I), B2M LOH, immunopeptidomics, human endogenous retroviruses (HERV), and microbiome, have been studied for their impact on the clinical response of patients with various types of cancer to ICI. Advanced computational methods that offer enhanced data integration through machine learning and/or deep learning have the potential to increase the clinical utility of these predictive tools. Stratifying cancer patients based on predicted therapeutic responses will guide the development of precision immunotherapy, providing the maximum clinical benefit while minimizing the risk of adverse events.
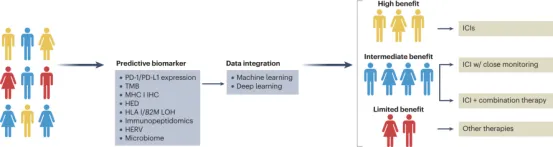
Figure 5. Antigen Presentation Can Be Associated With Clinical Response to ICI [2]
Immunopeptidomics facilitates the collective identification and quantification of HLA-presented peptides specific to samples, utilizing liquid chromatography (LC) and MS for immunopurification and analytical measurements. Predominantly exploratory in nature, the majority of immunopeptidomics research employs data-dependent acquisition (DDA) for MS, enabling the generation of detailed peptide MS/MS spectral fingerprints.
The standard immunopeptidomics workflow encompasses several steps. Initially, tumor tissues and normal tissues from cancer patients are processed to isolate HLA peptides. This process involves plate-based high-throughput sequencing (HTS) for HLA-I and HLA-II peptides, followed by immune-affinity purification and peptide extraction from the HLA complexes via reverse-phase C18 chromatography. The peptides are then analyzed via LC and introduced into a MS to acquire their MS/MS spectra information. Concurrently, DNA sequencing of tumor and adjacent non-tumor tissues is performed to pinpoint tumor-specific nonsynonymous mutations. Complementary RNA-seq and Ribo-seq analyses shed light on transcript expression and translationally active transcripts, respectively. This omics data informs the creation of a specialized protein reference database, selectively filtered to emphasize tumor-specific markers. This selection process involves the strategic use of existing genomic data to exclude markers common to healthy tissues while focusing on those prevalent in tumor samples. The curated reference database supports both the analysis of immunopeptidomics samples and the integration of public immunopeptidomics datasets for comprehensive MS analysis. Through MS database searching, both conventional and unconventional antigens are identifiable. Subsequent steps involve assessing the tumor specificity of these antigens and validating them via targeted MS techniques. Ultimately, the immunogenic potential of these peptides is subject to empirical testing. These peptides, particularly those that elicit an antigen-specific tumor response, are potential candidates for vaccine or for T-cell therapies, leveraging antigen-specific tumor-reactive T cells and corresponding TCRs for tumor immunotherapy.
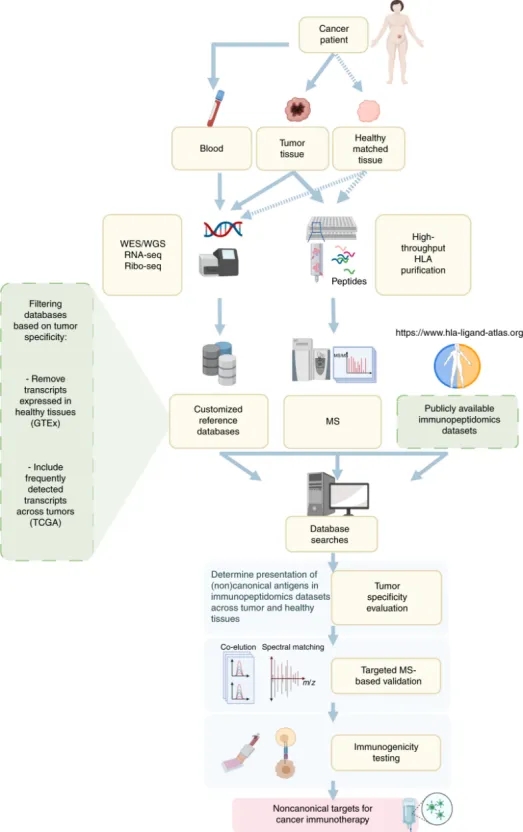
Figure 6. Immune Peptideome Flow [5]
1. Discovery of TAAs
Utilizing a variety of high-throughput multi-omics analysis techniques, including Next-Generation Sequencing (NGS) and MS/MS, enables the discovery and characterization of TAAs. These methodologies form two distinct approaches of antigen discovery: computational predictions of presumed HLA-presenting antigenic peptides based on HLA-binding characteristics, and direct identification of naturally presented HLA-eluted ligands with annotations in protein sequence databases.
(1) Prediction of New Antigens and Their Binding Capabilities via Bioinformatics.
Key bioinformatics analyses in the workflow for new antigen characterization encompass analyzing patient sequences to ascertain HLA types, alongside predicting the corresponding major histocompatibility complex (MHC) for each tumor. This involves examining various types of somatic mutations, including single nucleotide variants (SNVs), and assessing deletions, insertions, and fusions. It also includes analyzing the peptides for their predicted expression, processing, and binding capabilities to the patient's MHC. Following this, candidate vaccines are designed, and additional analyses are undertaken to evaluate T-cell responses.
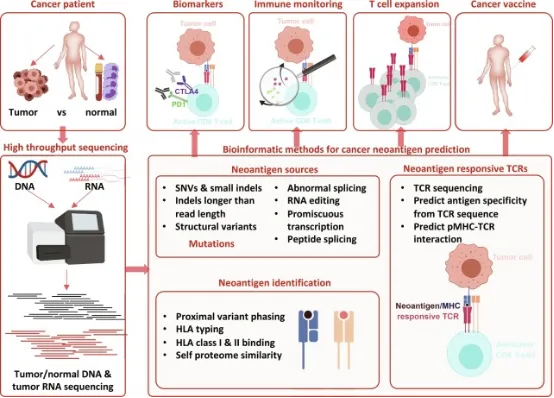
Figure 7. Overview of Bioinformatics Characterization of Neoantigens [6]
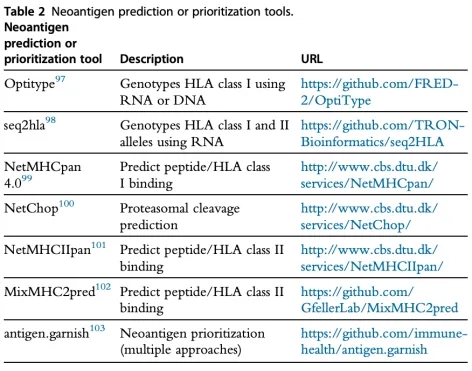
Figure 8. Neoantigen Prediction Tool [6]
(2) Direct Identification of New Antigens via MS
Peptides are separated from the HLA complex through HLA-I and HLA-II immune affinity purification and reverse-phase C18 extraction. These peptides are then subjected to LC and introduced into a MS to acquire their MS/MS spectra information. While most immunopeptidomics studies are discovery-focused, employing DDA MS methods, the data-independent acquisition (DIA) technique enhances peptide identification rates and significantly improves peptide reproducibility and quantitation across multiple samples. However, the implementation of DIA in immunopeptidomics to date requires a spectral library, restricting the discovery of novel altered or atypical peptides. Targeted MS has been applied in various manners within immunopeptidomics, serving as the most reliable method for tracking specific ions in complex peptide mixtures and quantifying the abundance and copy numbers of cell surface-specific antigens.
For peptide identification, several database search algorithms have been developed to evaluate the similarity between spectra from given reference databases and MS/MS spectra acquired. Alternatively, de novo sequencing algorithms allow for direct reconstruction of sequences from MS/MS spectra.
The rapid advancement of next-generation DNA sequencing, RNA sequencing (RNA-seq), and ribosome sequencing (Ribo-seq) technologies has given rise to a new field known as proteogenomics. Proteogenomics aims to leverage genomic, transcriptomic, and translatomic data to enhance the interpretation of MS-based proteomic or immunopeptidomic data. This approach allows for the use of protein sequence databases that are general, partially, or fully tailored to the needs of cancer patients. The proteogenomic framework consists of three main components: database creation, peptide identification, and post-processing and validation procedures. Database creation can range from simple, universal databases to more complex, customized databases utilizing available sequencing data. Immunopeptidomic raw data can be either specifically generated for research purposes or sourced from public repositories. Peptide identification employs MS search tools, including database-dependent searches and de novo algorithms. Post-processing and validation strategies incorporate MS-based targeted validation, peptide feature assessment, HLA binding predictions, and further analyses to refine the identification of tumor-specific targets.
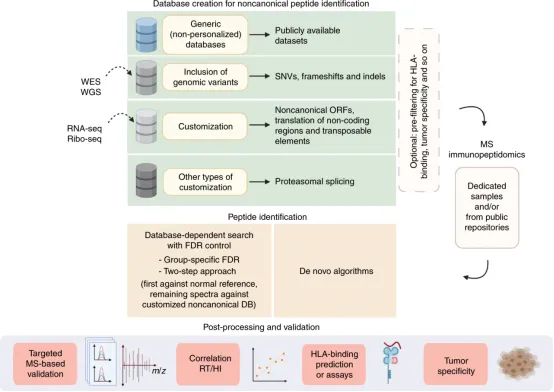
Figure 9. Computational Methods for Directed Immunopeptidomic Analysis of Protein Genomics [5]
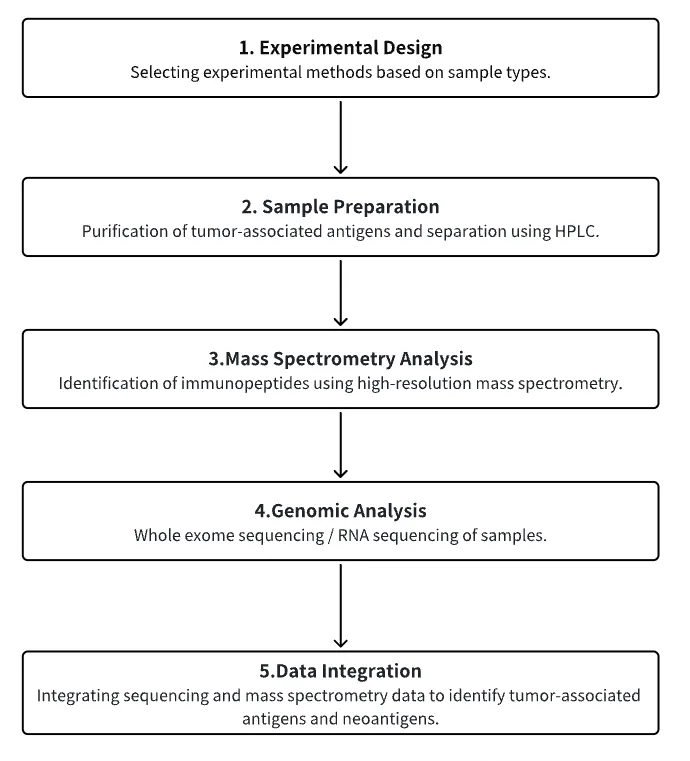
Analysis Workflow
1. Selection of Experimental Methods Based on Experimental Requirements
2. Purification and Separation of TAAs
3. Immunopeptidomics MS Analysis
4. Whole Exome Sequencing/RNA-Seq
5. Data Integration Analysis
Service Advantages
1. High-Throughput Sample Processing with Efficient Immunopeptide Enrichment
2. Reliable and Precise MS Analysis
3. Integrated Bioinformatics Analysis Across Genomics, Peptidomics, and Other Fields
Sample Results
1. Proteogenomic Analysis: RNA as a Source for Novel Tumor Antigen Identification
A comprehensive pan-cancer analysis may illuminate crucial commonalities linked to cancer immunogenicity and patient prognosis. A study encompassing 25 patients across 32 tumor types delivered an extensive multi-omics dataset aimed at novel antigen discovery through proteogenomics. Utilizing refined computational strategies, it unveiled a plethora of tumor-specific and associated antigens. This research integrates DNA and RNA sequencing with MS-based immunopeptidomics of tumor specimens, assessing their immunogenicity and conducting a thorough validation process. In most patients exhibiting partial immunogenicity, the study identified multiple non-canonical HLA-binding peptides, validating 32 potential novel antigen candidates. The majority of these candidates were traced back to mutations found within the RNA dataset, highlighting RNA's significance as a cancer antigen source. This investigation underscores the importance of RNA-centered mutation detection in identifying shared biomarkers and potentially relevant novel antigen candidates.
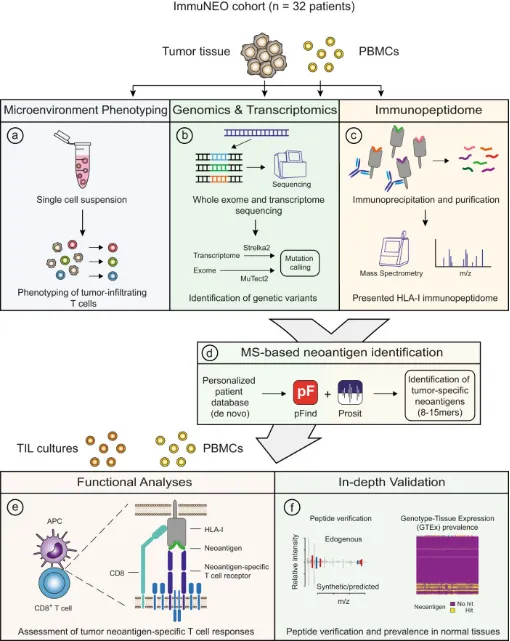
Figure 10. Computational Method for Protein Genomics-Directed Immunopeptides Analysis [7]
2. Novel Antigen Identification in Immunopeptidomics with Differential Ion Mobility Mass Spectrometry (DIM-MS)
Understanding the properties of HLA peptides, or immunopeptides, is essential for precision cancer medicine, though direct identification from clinical tissue biopsies MS presents considerable technical challenges. To overcome these obstacles, research introduced high-field asymmetric waveform ion mobility spectrometry (FAIMS), utilizing DIM-MS for efficient gas-phase fractionation, ideal for rare samples. Establishing DIM-MS for immunopeptidomics analysis, the study managed to analyze an average of 9.17 mg of normal and tumor colorectal tissue from the same patients (n=42), identifying an average of 4,921 immunopeptides. Within these 44,815 unique immunopeptides, two novel antigens, KRAS-G12V and CPPED1-R228Q, were identified. These antigens were confirmed through targeted MS in parallel reaction monitoring (PRM) mode using synthetic peptides. Comparative analysis of individual immunopeptides revealed tumor-specific processing, indicating that many more potential novel antigens remain unidentified directly from tumor tissues. Subsequently, the study screened cell lines with known oncogenic KRAS mutations, identifying two additional novel antigens carrying KRAS-G12V. These findings demonstrate the efficacy of the established FAIMS-assisted DIM-MS in identifying immunopeptides and potential novel antigens directly from limited samples, such as clinical tissues.
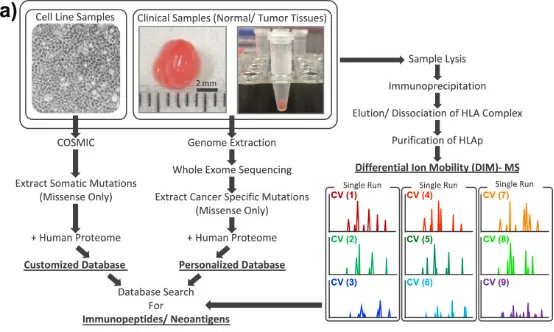
Figure 11. Proteomic Workflow for Personalized Immunopeptiomics Analysis [8]
Naïve MS screening is applied to discover SARS-CoV-2 replication inhibitors. After incubating the receptor with a mixture of potential ligands in a low molar concentration volatile aqueous buffer (such as 3 mM ammonium acetate), the desorbed receptor-ligand complexes are measured using ESI-MS on an ultra-high-resolution MS. During the ESI-MS analysis, high mass receptor ions are detected as a series of multiply charged species (black), while the ligand-receptor complexes appear as a series of slightly higher m/z value multiply charged peaks, corresponding to the receptor plus the attached ligand (red).
3. Formulation of Personalized Multi-neoantigen Vaccines for Breast Cancer
Harnessing the immune system to purposefully identify and eradicate tumors marks a significant advancement in clinical oncology. Nonsynonymous mutations (neoantigen peptides) have been pinpointed as potent targets for cancer treatment. This insight is pivotal for enhancing active immunotherapies, such as cancer vaccines, because neoantigen-specific T cells are not compromised by immune tolerance mechanisms, nor do they inflict damage on healthy tissue. A study has successfully integrated MS-based immunopeptidomics with whole exome sequencing to pinpoint mutations in mouse breast cancer cells. Subsequently, neoantigen peptides were synthesized using a copper-free click chemistry approach and covalently bonded with virus-like nanoparticles, culminating in the development of a vaccine against mouse breast cancer.
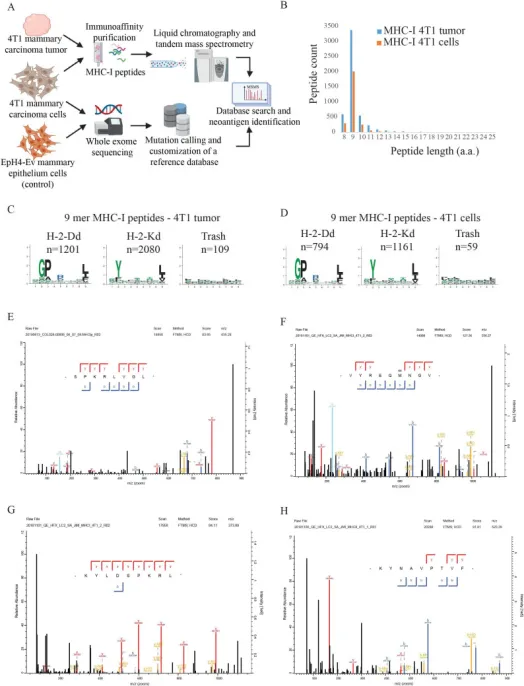
Figure 12. Identification of Tumor Neoantigens [9]
4. Proteogenomic Discovery of Neoantigens Aids in Personalized Multi-antigen Targeted T Cell Immunotherapy for Brain Tumors.
The identification of neoantigens in pediatric brain tumors is impeded by their low mutational load and limited tissue availability. Research has formulated a proteogenomic method that merges tumor DNA/RNA sequencing with MS proteomics to detect tumor-restricted (neoantigen) peptides spawned by numerous genomic alterations. This approach facilitates the creation of highly specific, autologous, personalized T cell immunotherapy. Evidence suggests that aberrant splicing junctions are the principal source of neoantigens in medulloblastoma, a prevalent form of pediatric brain tumor. Proteogenomics has managed to identify tumor-specific peptides that are immunogenic and trigger MHC II-based T cell responses. Moreover, polyclonal and multifunctional T cells aimed at tumor-specific peptides have demonstrated efficacy in eradicating tumor cells in vitro. Focusing on TSAs circumvents the challenges of central immune tolerance while potentially offering a safety margin that enhances the compatibility with other immune-activating treatments. These discoveries underscore the significance of proteogenomic identification of immunogenic tumor-specific peptides and establish a groundwork for personalized targeted T cell therapies for children suffering from brain tumors.
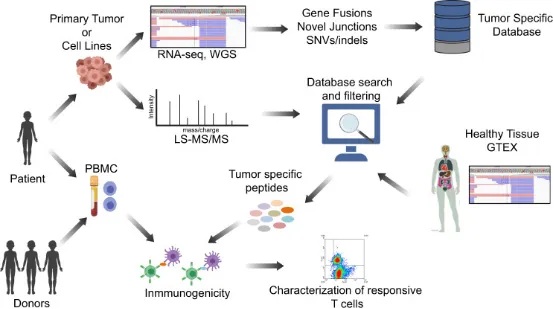
Figure 13. Identification of Tumor Neoantigens [10]
Sample Submission Requirements
1. Provision of pre-purified samples to the greatest extent possible.
2. Minimization of impurity contamination.
Services at Mtoz Biolabs
1. Complete Experimental Procedures
2. Relevant Instrument Parameters
3. Raw Experimental Data
4. Immunopeptide Data Analysis Report (including information on TAAs, neoantigens, etc.)
Applications
1. Precision Cancer Immunotherapy through Targeting Neoantigens
Immunotherapeutic strategies that target cancer neoantigens are recognized for their safety, efficacy, and precision. The identification of neoantigens is predominantly achieved through advanced genomic technologies, including next-generation and high-throughput single-cell sequencing; proteomic approaches such as MS and the application of bioinformatics tools that analyze HTS and MS data alongside biological databases. Therapeutic interventions related to neoantigens, such as vaccines, CD8+ and CD4+ T cells that are specific to neoantigens, and dendritic cells pulsed with neoantigens, have gained widespread clinical application. Furthermore, neoantigens act as biomarkers for assessing responses to immunotherapy, resistance mechanisms, and prognostic outcomes. Treatments based on neoantigens constitute a critical and promising branch within the field of cancer immunotherapy.
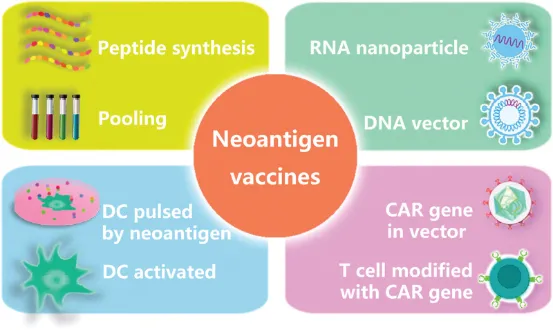
Figure 14. Neoantigen-Based Therapies [11]
2. Establishment of a Tumor Neoantigen Database
The capacity of neoantigens to evoke T cell responses and precipitate tumor regression underscores the expansive potential of immunotherapy. Recent advancements have seen the dbPepNeo database upgraded to version 2.0, which now encompasses approximately 801 high-confidence neoantigens, marking a 170% increase, and 842,289 low-confidence HLA immunopeptides, up 107%. Significantly, this version introduces for the first time 55 class II HC neoantigens and 630 neoantigen-reactive T cell receptor β (TCRβ) sequences. In addition, the development of two innovative analytical tools, DeepCNN-Ineo and BLASTdb, enhances the database's utility. DeepCNN-Ineo is designed to predict the immunogenicity of class I neoantigens, while BLASTdb facilitates local alignment searches for sequence similarities within dbPepNeo2.0.
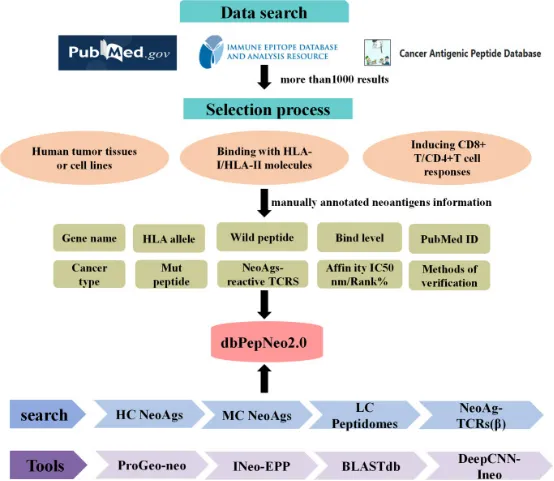
Figure 15. Content and Structure of dbPepNeo2.0 Database [12]
FAQ
Q1: How to enrich TAAs?
Peptides presented by HLA are either non-specifically dissociated through mild acid elution (MAE) or purified HLA: peptide complexes using specific antibodies through immunoprecipitation (IP), followed by the separation of peptides and HLA molecules. Compared to MAE, IP sample preparation represents the current preferred method due to its high specificity and higher yield.
Q2: What data acquisition methods are chosen for immunopeptidomics MS identification?
Different MS acquisition methods have been applied to the detection of the immunopeptidome and the identification of tumor antigens: DDA, Targeted Data Acquisition, and the more recent DIA. Since most immunopeptidomics experiments aim to identify unknown epitopes, discovery-driven DDA is the most commonly used method for generating tandem mass spectra. The acquired data can be used for the identification of peptides and their amino acid sequences, either by comparing them to theoretical spectra of all precursors with a similar mass-to-charge ratio (m/z) in a database, by resequencing, or a combination of both methods. DDA allows the identification of thousands of unique class I HLA-presented peptides. However, because the heuristic selection of peptide precursors and subsequent fragmentation depend on the abundance of peptide precursors, low abundance peptides (such as most tumor-specific epitopes) are easily missed by DDA, which also lacks repeatability and quantitative accuracy. In contrast, DIA offers improvements in reproducibility and quantitation. In DIA, all precursors within defined mass-charge windows are fragmented in an unbiased manner, producing highly convoluted spectra. This process is repeated across the entire mass-charge range to generate a "digital map" of the analyzed sample. To identify peptides from these spectra, they need to be compared to a spectral library containing fragment and retention time information of relevant peptides. Generating high-quality spectral libraries typically requires extensive DDA analysis of the same or related samples beforehand.
References
[1] Peri, A., Salomon, N., Wolf, Y. et al. The landscape of T cell antigens for cancer immunotherapy. Nat Cancer 4, 937–954 (2023). https://doi.org/10.1038/s43018-023-00588-x.
[2] Yang K, Halima A, Chan TA. Antigen presentation in cancer - mechanisms and clinical implications for immunotherapy. Nat Rev Clin Oncol. 2023 Sep;20(9):604-623. doi: 10.1038/s41571-023-00789-4. Epub 2023 Jun 16. PMID: 37328642.
[3] Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discov. 2022 Apr;21(4):261-282. doi: 10.1038/s41573-021-00387-y. Epub 2022 Feb 1. PMID: 35105974; PMCID: PMC7612664.
[4] Blass, E., Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 18, 215–229 (2021). https://doi.org/10.1038/s41571-020-00460-2.
[5] Chong C, Coukos G, Bassani-Sternberg M. Identification of tumor antigens with immunopeptidomics. Nat Biotechnol. 2022 Feb;40(2):175-188. doi: 10.1038/s41587-021-01038-8. Epub 2021 Oct 11. PMID: 34635837.
[6] Boegel S, Castle JC, Kodysh J, O'Donnell T, Rubinsteyn A. Bioinformatic methods for cancer neoantigen prediction. Prog Mol Biol Transl Sci. 2019;164:25-60. doi: 10.1016/bs.pmbts.2019.06.016. Epub 2019 Jul 18. PMID: 31383407.
[7] Tretter C, de Andrade Krätzig N, Pecoraro M, Lange S, Seifert P, von Frankenberg C, Untch J, Zuleger G, Wilhelm M, Zolg DP, Dreyer FS, Bräunlein E, Engleitner T, Uhrig S, Boxberg M, Steiger K, Slotta-Huspenina J, Ochsenreither S, von Bubnoff N, Bauer S, Boerries M, Jost PJ, Schenck K, Dresing I, Bassermann F, Friess H, Reim D, Grützmann K, Pfütze K, Klink B, Schröck E, Haller B, Kuster B, Mann M, Weichert W, Fröhling S, Rad R, Hiltensperger M, Krackhardt AM. Proteogenomic analysis reveals RNA as a source for tumor-agnostic neoantigen identification. Nat Commun. 2023 Aug 2;14(1):4632. doi: 10.1038/s41467-023-39570-7. PMID: 37532709; PMCID: PMC10397250.
[8] Minegishi Y, Kiyotani K, Nemoto K, Inoue Y, Haga Y, Fujii R, Saichi N, Nagayama S, Ueda K. Differential ion mobility mass spectrometry in immunopeptidomics identifies neoantigens carrying colorectal cancer driver mutations. Commun Biol. 2022 Aug 18;5(1):831. doi: 10.1038/s42003-022-03807-w. PMID: 35982173; PMCID: PMC9388627.
[9] Mohsen MO, Speiser DE, Michaux J, Pak H, Stevenson BJ, Vogel M, Inchakalody VP, de Brot S, Dermime S, Coukos G, Bassani-Sternberg M, Bachmann MF. Bedside formulation of a personalized multi-neoantigen vaccine against mammary carcinoma. J Immunother Cancer. 2022 Jan;10(1):e002927. doi: 10.1136/jitc-2021-002927. PMID: 35017147; PMCID: PMC8753436.
[10] Rivero-Hinojosa S, Grant M, Panigrahi A, Zhang H, Caisova V, Bollard CM, Rood BR. Proteogenomic discovery of neoantigens facilitates personalized multi-antigen targeted T cell immunotherapy for brain tumors. Nat Commun. 2021 Nov 18;12(1):6689. doi: 10.1038/s41467-021-26936-y. PMID: 34795224; PMCID: PMC8602676.
[11] Zhang Q, Jia Q, Zhang J, Zhu B. Neoantigens in precision cancer immunotherapy: from identification to clinical applications. Chin Med J (Engl). 2022 Jun 5;135(11):1285-1298. doi: 10.1097/CM9.0000000000002181. PMID: 35838545; PMCID: PMC9433083.
[12] Lu M, Xu L, Jian X, Tan X, Zhao J, Liu Z, Zhang Y, Liu C, Chen L, Lin Y, Xie L. dbPepNeo2.0: A Database for Human Tumor Neoantigen Peptides From Mass Spectrometry and TCR Recognition. Front Immunol. 2022 Apr 13;13:855976. doi: 10.3389/fimmu.2022.855976. PMID: 35493528; PMCID: PMC9043652.
How to order?

