





Hydrogen Deuterium Exchange Mass Spectrometry, HDX MS Service
Hydrogen deuterium exchange mass spectrometry (HDX MS) represents a sophisticated technique in mass spectrometry for investigating the spatial conformation of proteins, extensively employed across academia and the biopharmaceutical industry for the analysis of protein structures. The main principle of this technique is that when the protein is placed in a heavy aqueous solution, the hydrogen atoms within the protein's amide backbone can interchange with the environmental deuterium atoms, and exhibits a higher hydrogen exchange rate when exposed to solvents or within regions of conformational activity, while the hydrogen exchange rate is lower in the core structural regions. Consequently, the rate of hydrogen-deuterium exchange in peptide fragments generated through the enzymatic digestion of proteins in various states can be methodically compared. This allows for the analysis of proteins advanced structures, dynamic alterations, sites of protein-protein interactions, epitopes, conformations, and modifications in protein-ligand binding structures. HDX MS offers the benefits of requiring minimal sample sizes and enable the direct examination of proteins' natural conformations in solution.
In hydrogen deuterium exchange mass spectrometry (HDX MS), owing to the underlying principle of hydrogen-deuterium atom exchange, the back-exchange of hydrogen-deuterium atoms significantly impacts the precision and accuracy of experimental results, even at a temperature of 0°C, amide hydrogen exchange proceeds with a half-life ranging from 30 to 120 minutes, necessitating that peptides be introduced into the mass spectrometer within 10 minutes, with stringent control over temperature and pH within the range minimizing back-exchange reaction rates to minimize the back-exchange of hydrogen-deuterium atoms as much as possible.
Initially, the protein undergoes incubation in a deuterium buffer subsequently, over a series of time points enabling deuterium atoms to incorporate into the protein backbone, the exchange reaction is halted by transitioning to an acidic pH and reducing the temperature, subsequently, acidic functional proteases digest the protein, following desalting, the digested products are separated using a UPLC system and transported to a mass spectrometer, where the peptides are ionized by electrospray and mass analysis to determine the mass increase due to deuterium uptake. By plotting the deuterium exchange profiles of the polypeptide at various time points or comparing the experimental group with the control group, we can analyze the structural alterations of the protein in specific regions, the details are as follows:
Experimental Instruments
1. HDX MS Analysis System ( nanoACQUITY UPLC HD-Exchange System)
2. Electrospray-Combined Orbitrap Mass Spectrometer( Q Exactive™ Hybrid Quadrupole-Orbitrap™ Mass Spectrometer, Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer)
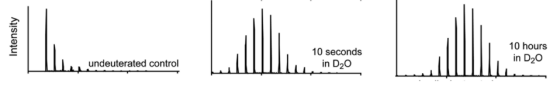
Figure 1. Mass Shifts Due to Different Time Stamps in HDX MS
Analysis Workflow
1. Deuterium Labeling
Proteins are incubated in deuterium buffer and labeled time points are designed.
2. Quenching
Add quenching solution and adjust it to pH2.5, 0℃, and reduce the exchange rate to a minimum.
3. Enzymatic Hydrolysis
Use acid protease to cut protease into peptides, such as pepsin.
4. LC-MS
The deuterated peptides are separated by low-temperature HPLC/UPLC, and then entered into the mass spectrometry by electrospray ionization, and the deuterated value is monitored according to the mass shift of the peptide.
5. Data Processing
Combined with the software MassLynx®, DynamX HDX, and ProteinLyns Global SERVERTM, automated, visualized, and interactive result processing is carried out.
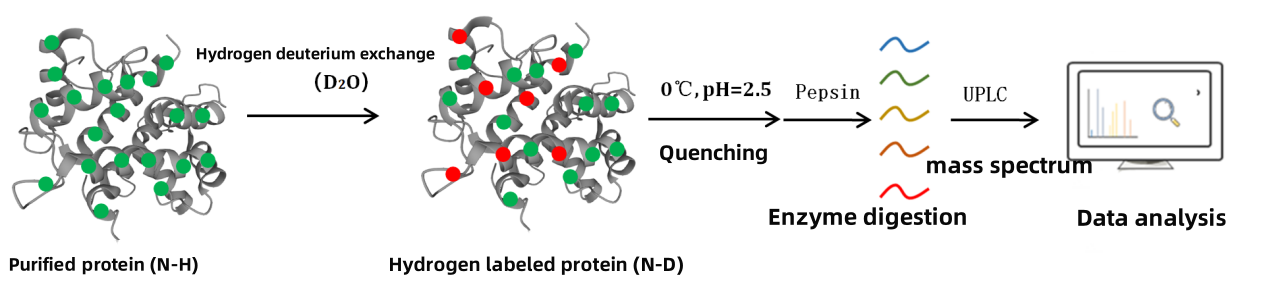
Figure 2. HDX MS Research Process
Services Advantages
1. MtoZ Biolabs uses waters' advanced nano ACQUITY UPLC HD-Exchange System to ensure the accuracy of the experiments.
2. According to the customer's target protein, the enzyme digestion combination is designed to obtain comprehensive peptide coverage.
3. Compared with ordinary large-particle packing materials, the sub-nanometer chromatography particle packing material can greatly shorten the requirements of liquid phase analysis time without losing the chromatographic separation effect, and has ultra-high resolution.
4. The on-line automatic pretreatment and high-resolution mass spectrometer are used , which not only ensures the reliability of the experimental data, but also improves the experimental efficiency, and fully guarantees the reliability of the pretreatment effect and identification results.
5. A variety of data analysis softwares are used to provide you with more comprehensive and sufficient identification results.
Sample Results
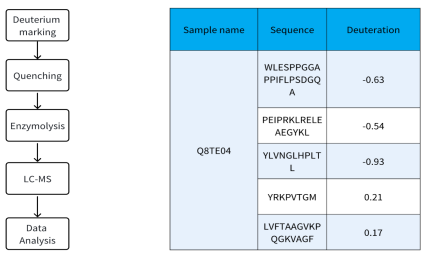
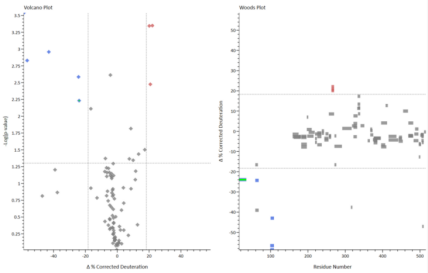
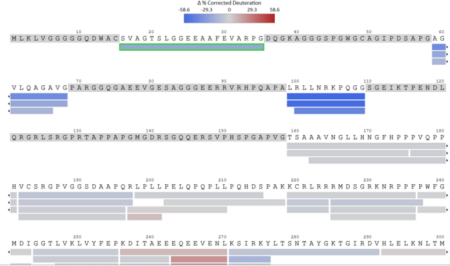
Sample Submission Requirements
1. Protein-ligands and protein samples.
2. Sample concentration: >1 μg/μl.
3. Sample volume: >10 μg.
4. To ensure accurate identification, maximize the purity of your samples.
5. Note: Samples should preferably be free of detergent.
Services at MtoZ Biolabs
Following our team's hydrogen-deuterium exchange mass spectrometry analysis, you will observe the spatial conformational changes of your target protein at various time points, and its interaction with the ligand in comparison to its native state. Concurrently, we will furnish you with the pertinent experimental protocols, mass spectrometry parameters, total ion chromatograms, raw mass spectrometry data, as well as detailed analyses of protein spatial epitopes and conformational data.
Applications
1. Spatial conformation and active site studies of proteins.
2. Dynamic changes in protein spatial epitopes.
3. Effect of changes in protein structure on antibody binding.
4. Protein-protein interaction site studies.
5. Combined with crystallography methods to systematically study protein structure.
6. Subtypic structure studies of pharmaceutical compounds.
FAQ
Q1: What forms will the structural changes take as a result?
In the results, in addition to presenting the deuterium levels for each peptide within the protein, we offer a range of visual representations derived from the deuterium levels, such as the butterfly plot that concurrently displays the deuterium levels across all groups within the HDX MS dataset, integrating multiple time points within a singular visualization, enabling the tracking of alterations throughout the complete temporal span of the experiment. The Woods plot illustrates the deuteration levels across all sequences, and the peptides' length and positions. The volcano plot additionally presents the P-values, enabling rapid identification of sequences exhibiting significant deuteration changes among the peptides. Furthermore, mapping deuterium values to the 3D structure of the deuterated protein can elucidate conformational changes at specific sites of protein more directly.
Q2: How to ensure the reliability of experimental results?
Hydrogen deuterium exchange mass spectrometry has certain requirements for the purity of the sample, please purify the protein as much as possible when providing the sample, and design biological replicates or technical replicates according to the experiment. More importantly, it is necessary to determine the appropriate exchange time, and in order to obtain more comprehensive and accurate information, it is recommended that the marking time spans four orders of magnitude: 0.1 min, 1 min, 10 min and 100 min.
How to order?

