





Comprehensive Host Cell DNA (HCD) Detection Service
Several methodologies are employed for quantifying rDNA within biological processes. These include traditional methods (PicoGreen method, hybridization method, and threshold method) as well as more commonly used methods nowadays such as quantitative real-time PCR (qPCR), capillary electrophoresis (CE), and primer-based whole genome amplification (pWGA) technology.
1. PicoGreen Method
This method involves a dye named PicoGreen® that binds to double-stranded DNA, inducing fluorescence, in contrast to the minimal fluorescence emitted by unbound dye. The PicoGreen method is notably straightforward and cost-effective, suitable particularly for samples with relatively high DNA levels, such as those from upstream bioprocesses. Its major drawback is its low sensitivity, detecting only about 0.2 ng of pure DNA. Like all rDNA techniques, this method is also prone to interference and impacts from other components, especially in the presence of high concentrations of recombinant proteins. The primary limitation is its lack of sensitivity, as per the World Health Organization (WHO) guidelines, which stipulate that each dose of medication should not contain more than 10 ng of rDNA. Therefore, this approach is rarely suitable for detecting residual DNA in final products. However, if properly validated, it can be applicable to low-dose protein products, such as certain vaccines. Besides, the PicoGreen® assay offers advantages of high throughput and cost-effectiveness.
2. Hybridization Method
Another technique for quantifying rDNA is hybridization. Hybridization quantifies rDNA by utilizing probes tagged with radioactive or fluorescent markers to identify specific single-stranded DNA (ssDNA) sequences. These probes then hybridize with denatured DNA samples that are immobilized on a membrane. The quantity of DNA is estimated by comparing the signal intensity to DNA standards, typically genomic DNA from the host. The advantage of this technique is its similarity to the widely used Southern Blot in molecular biology, which requires no special training and is relatively low-cost. It also allows for the estimation of DNA size by running the DNA alongside standards of various molecular weights prior to blotting. However, similar to the PicoGreen® assay, its main drawback is low sensitivity (typically around 10 ng/mL) and longer assay time (about 48 hours). Due to these limitations, its use in the biotechnology industry has declined.
3. Threshold Method
This method utilizes ssDNA-binding proteins to bind and capture denatured single-stranded rDNA. Detection is performed using a monoclonal anti-DNA antibody linked to urease, with enzyme activity causing pH changes in a reaction chamber. In a reaction chamber, the enzymatic hydrolysis of urea results in changes in pH values. Consequently, the rate of pH change is directly proportional to the amount of DNA in the sample. The quantity of DNA is then calculated by comparing this change with the signals from DNA standards. The threshold method's strengths lie in its high sensitivity (1-3 pg) and standardized protocols, but it suffers from higher costs and limited throughput. Another consideration for this assay is the limitation on the detectable size of DNA fragments in biologics. The method relies on the binding of denatured DNA with ssDNA-binding proteins and monoclonal anti-DNA molecules, thus failing to detect DNA fragments smaller than 600 bp. During the biomanufacturing process, DNA can be sheared into smaller sizes through filtering and chromatography, limiting the applicability of this threshold-based method in the detection of host cell residual DNA.
4. qPCR Method
The detection of rDNA in biological product development necessitates a method that is both rapid and cost-effective, while also maintaining high sensitivity. In recent years, PCR-based technologies have emerged as the predominant methods for rDNA quantification in the biotechnology sector. In the PCR process, sequence-specific DNA templates are amplified to produce billions of copies within just 1-2 hours, enabling the detection of minuscule DNA quantities. The earliest PCR methods were based on end-product analysis, typically using agarose gel electrophoresis to evaluate the final PCR products. This additional step constrained the quantitative capabilities of the method. Significant advancements have been made in technologies that allow real-time monitoring of DNA amplification. Quantification of rDNA in bioproducts can be precisely achieved by measuring the PCR products during the early stages of reverse transcription -PCR (RT-PCR) amplification, due to a strong correlation between initial DNA levels and the signals detected during the early exponential phase. Quantifying DNA during the linear or plateau phases results in significant variability.
qPCR employs various sophisticated chemistries, three of which are particularly common. The first method involves the use of a double-stranded DNA-binding dye, such as SYBR® Green (provided by Life Technologies, Foster City, CA). When excited by light of an appropriate wavelength, SYBR® Green exhibits very low background fluorescence as a free dye. In contrast, when SYBR® Green binds to the minor grooves of the double-stranded DNA produced in the PCR reaction, the fluorescence output significantly increases, approximately 2,000 times stronger than the free dye, thus offering an excellent signal-to-noise ratio. The second approach involves dye-labeled primers designed with a quenching hairpin structure that, during PCR, unwinds and anneals to the DNA template, enhancing fluorescence substantially as it transitions from the hairpin to a linear structure. This technique allows for the simultaneous monitoring of multiple genes, enhancing its utility for complex assays. The third method employs TaqMan® technology, which uses a fluorescently labeled probe positioned between two sequence-specific primers. As PCR progresses, polymerase's exonuclease activity cleaves the probe, separating the fluorescent dye from the quencher and resulting in a marked fluorescence increase. Like SYBR® Green, the TaqMan® method provides a signal-to-noise ratio exceeding 1000, rendering it exceptionally sensitive and suitable for precise detection needs in biotechnological applications.
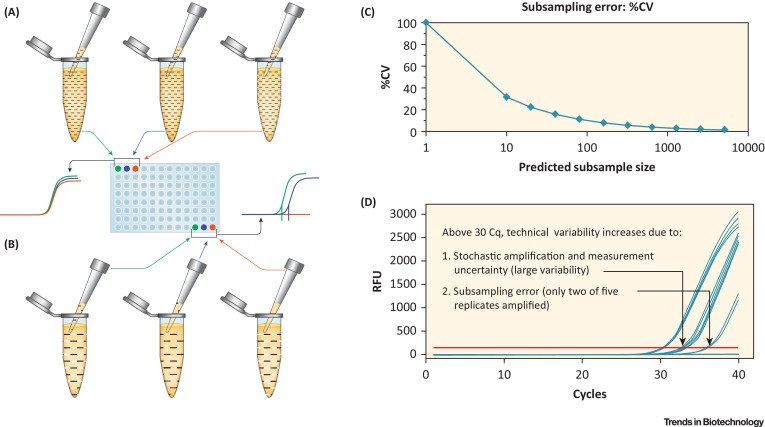
Figure 1. Schematic Diagram of DNA Quantitative PCR (qPCR) Data [1]
5. pWGA Method
Detecting DNA through hybridization assays, immunoassays, or real-time PCR offers distinct benefits but also presents inherent limitations. PCR-based detection primarily targets specific sequences that do not represent the entire genome; thus, it may fail to detect other sequences if the specified target sequence is absent. This is particularly important considering that biological processes can shear DNA into smaller fragments, and PCR can only detect rDNA if the target DNA sequence is present. Although hybridization and antibody/affinity-based DNA detection methods aim to identify all potential DNA sequences that may be present, these techniques have limited sensitivity because, unlike PCR, they do not involve amplification of DNA. Threshold®'s antibody/affinity technology is limited by sequence size. To address this, alternative technologies have been explored, one of which is WGA technology. WGA employs different amplification patterns and uses unique primers to enhance specificity. Similar to PCR, all WGA methods amplify small amounts of DNA to achieve the desired sensitivity.
The initial WGA system was PCR-based DNA amplification system, where specific DNA sequences are added to the ends of all DNA fragments using a DNA ligase, and these sequences serve as starting points for PCR amplification. Through this method, all DNA fragments are amplified using PCR with primers that bind to the added sequences. When amplified in a real-time quantitative PCR setup, this allows for the detection of rDNA without sequence bias or selectivity. Thus, compared to standard PCR techniques, this WGA system offers greater selectivity and sensitivity in detecting rDNA in biologics. However, the limitation of this technique lies in the efficiency of adding specific DNA sequences to all DNA fragments, as fragments without these sequences will not be amplified by the PCR scheme and thus remain undetected.
The second type of WGA system aims to overcome the DNA sequence requirements inherent in PCR techniques by using degenerate (less specific) DNA primers. Theoretically, these degenerate primers can bind to a broader range of total DNA sequences, thereby providing relatively high uniformity in DNA amplification across the entire genome and minimizing sequence bias. Additionally, this technology enhances rDNA detection by reducing the sequence preference often encountered in standard PCR protocols.
The third WGA method employs the Escherichia coli DNA replication mechanism, substituting Taq DNA polymerase with primase and incorporating DNA helicase. The use of primase eliminates the need for traditional primers to initiate DNA amplification. The presence of DNA helicase allows double-stranded DNA targets to unwind at 37°C, thus enabling DNA amplification to proceed at the same temperature. Additionally, the presence of primase provides multiple initiation sites, facilitating more efficient amplification of target DNA.
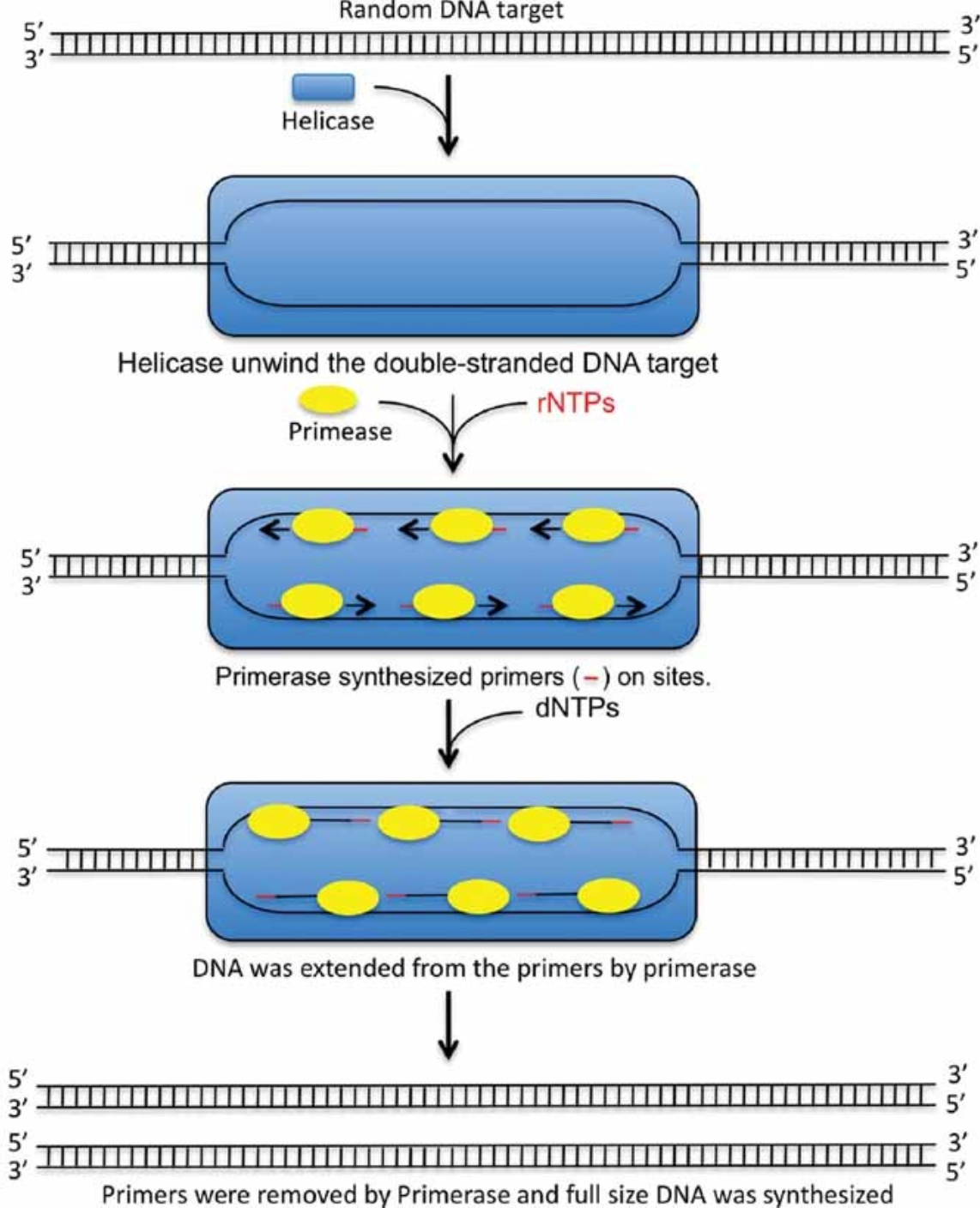
Figure 2. Whole Genome Amplification Model of pWGA System [2]
Analysis Workflow
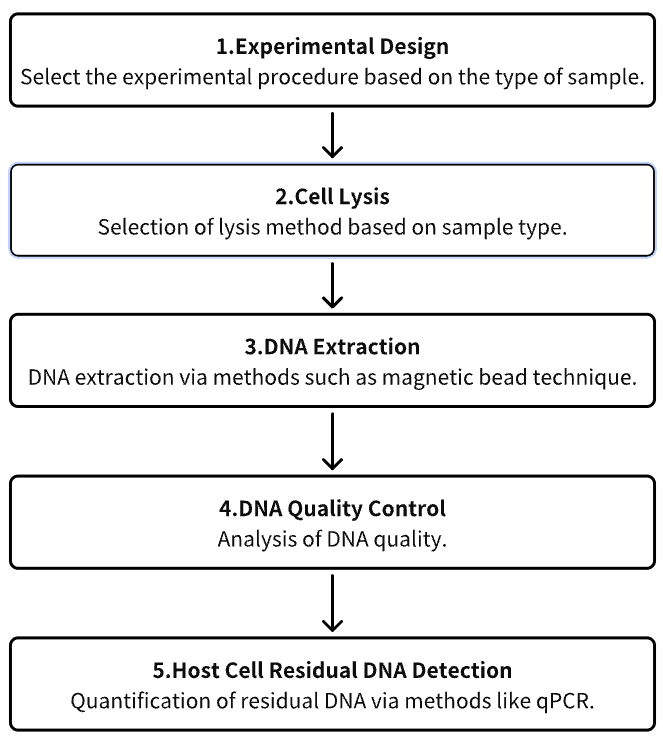
1. Experimental Procedure Determination Based on Requirements
2. Cell Lysis
3. DNA Extraction
4. DNA Quality Control
5. Host Cell Residual DNA (HCD) Detection
Service Advantages
1. High Credibility Identification and Characterization of HCD
2. Automated High-Throughput DNA Extraction Process
3. Effective DNA Detection System and Extensive Testing Experience
Sample Results
1. Development and Characterization of a New HCD Detection Method Using Ultra-Sensitive Fluorescent Nucleic Acid Dye “PicoGreen”
A novel research has been developed for HCD detection using PicoGreen, capable of identifying short double-stranded DNA (ds-DNA) in cell culture supernatants and processing intermediates. The PicoGreen DNA assay was evaluated by determining its detection limits for various DNA lengths, observing short ds-DNA presence in cell culture supernatants and process intermediates, assessing dose-dependence, and comparing it against traditional assays for HCD quantification in actual samples. PicoGreen DNA detection can identify ds-DNA as short as 20 bp, and its sensitivity is comparable to that of the Threshold system, which involves additional steps of SDS/proteinase K digestion and DNA concentration. However, compared to the results of PicoGreen DNA detection, the Threshold system generally underestimates the amount of DNA identified in cell culture supernatants and process intermediates. PicoGreen DNA detection not only offers higher accuracy but also provides a simpler procedure than the traditional methods using the Threshold system, particularly for measuring HCD levels in cell culture supernatants and process intermediates. This newly developed DNA detection method is expected to stand out in the detection of HCD, especially in biopharmaceutical applications.
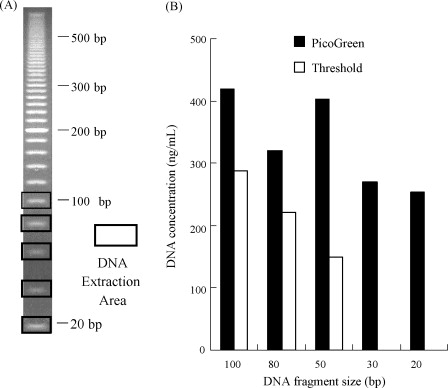
Figure 3. Detection of Extracted DNA Using Threshold System and PicoGreen Method [3]
2. qPCR Detection of CHO Host Cell DNA
Hamster ovary (CHO) cells are widely used in the production of recombinant proteins for human therapeutics. The paper have proposed a cost-effective and sensitive qPCR detection method for the presence of residual DNA from hamsters (Cricetulus griseus). This method is suitable for high-throughput screening using a 96-well plate format. The qPCR primers are designed to amplify a 150 bp region of the hamster genomic DNA. The specificity of the probes was evaluated using genomic DNA from mouse fibroblast lines, human kidney cell lines, and hamster ovary cell lines as templates. The sensitivity of the qPCR in detecting the genomic DNA of the hamster cell line CHO DG44 was also compared. These primers can be used in qPCR to detect the contamination of hamster DNA in purified protein samples, with sensitivity as low as 300 fg of genomic DNA.
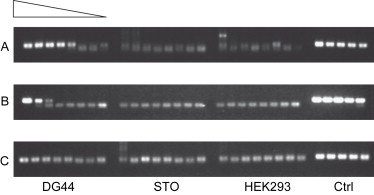
Figure 4. PCR specificity tests show that the primers can detect the presence of hamsters, but cannot detect mouse and human genomic DNA. [4]
3. Comparative Analysis of Residual Host Cell DNA Detection Performance in Viral Vaccines Produced in Vero Cells
Residual host cell DNA (rcDNA) in biopharmaceutical production poses significant safety concerns. Over time, various methods such as hybridization assays, Threshold assays, and qPCR have been employed for rcDNA quantification. Research has compared internal detection methods for quantifying rcDNA in virus vaccines produced in Vero cells. The Vero α-satellite sequence qPCR was compared with the hybridization method and threshold method in terms of specificity, sensitivity, and precision. Furthermore, the study also evaluated the effect of using β-propiolactone (BPL) to inactivate viruses on rcDNA during the vaccine production process. The research demonstrated that the measured quantity of rcDNA is influenced by the analytical method used. Results indicated that the Vero cell DNA-specific qPCR analysis method has a broad dynamic range and is free from matrix interference across a range of products. Compared to the hybridization and threshold methods, the qPCR method offers higher sensitivity and specificity. The Vero α-satellite sequence qPCR is a specific and sensitive method for assessing the quantity of Vero rcDNA in highly purified vaccines.

Figure 5. β -actin Gene Sequence of Vero Resizing Fragment and the Position of Forward and Reverse Primers [5]
Sample Submission Requirements
1.Minimize Impurity Contamination
Services at MtoZ Biolabs
1. Complete Experimental Procedure
2. Relevant Instrument Parameters
3. Original Experimental Data
4. Data Analysis Submission
Applications
1. Development of a Standard Detection Method
A novel Vero cell DNA reference has been developed to standardize dot blot hybridization, which is extensively employed to quantify residual DNA in viral vaccines produced with Vero cells. The extraction of high-purity Vero cell DNA was followed by its characterization through Hind III enzyme digestion and subsequent DNA sequencing. The concentration of the bulk Vero cell DNA reference solution was accurately determined via collaborative calibration, yielding a concentration of 64.0±1.9 µg/mL with an OD260/OD280 ratio of 1.87, indicative of high DNA purity. This solution was then diluted to 40 ng/mL using a Tris-EDTA buffer containing bovine serum albumin as a lyoprotectant to enhance stability. The diluted reference was aseptically filled into vials (0.5 mL each) using an industrial-grade filler and was securely heat-sealed under an inert nitrogen atmosphere. The sensitivity of this Vero cell DNA reference was established at 1 to 5 pg/dot, demonstrating robust stability even after rigorous accelerated degradation testing. The successful establishment of a reference for quantifying Vero cell DNA will facilitate the standardization of dot blot hybridization for detecting residual host cell DNA.
References
[1] Taylor SC, Nadeau K, Abbasi M, Lachance C, Nguyen M, Fenrich J. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 2019 Jul;37(7):761-774. doi: 10.1016/j.tibtech.2018.12.002. Epub 2019 Jan 14. PMID: 30654913.
[2] Wang X, Morgan DM, Wang G, Mozier NM. Residual DNA analysis in biologics development: review of measurement and quantitation technologies and future directions. Biotechnol Bioeng. 2012 Feb;109(2):307-17. doi: 10.1002/bit.23343. Epub 2011 Nov 6. PMID: 21956148.
[3] Ikeda Y, Iwakiri S, Yoshimori T. Development and characterization of a novel host cell DNA assay using ultra-sensitive fluorescent nucleic acid stain "PicoGreen". J Pharm Biomed Anal. 2009 May 1;49(4):997-1002. doi: 10.1016/j.jpba.2009.01.022. Epub 2009 Jan 24. PMID: 19217735.
[4] Nissom PM. Specific detection of residual CHO host cell DNA by real-time PCR. Biologicals. 2007 Jun;35(3):211-5. doi: 10.1016/j.biologicals.2006.09.001. Epub 2006 Oct 30. PMID: 17071102.
[5] Vernay O, Sarcey E, Detrez V, Abachin E, Riou P, Mouterde B, Bonnevay T, Mallet L. Comparative analysis of the performance of residual host cell DNA assays for viral vaccines produced in Vero cells. J Virol Methods. 2019 Jun;268:9-16. doi: 10.1016/j.jviromet.2019.01.001. Epub 2019 Jan 4. PMID: 30611776.
How to order?

