





Global Phosphoproteomics Service
- Phosphorylation site identification
- Differential phosphorylation quantitation
- Signaling pathway enrichment analysis
- Kinase-substrate network prediction
- Identified over 1,000 exercise-regulated phosphorylation sites.
- Revealed an AMPK-centric kinase network and discovered novel AMPK substrates involved in energy metabolism, signal transduction, and cytoskeletal remodeling.
The global phosphoproteomics service is an integrated solution that combines tyrosine phosphorylation-specific antibody enrichment techniques with conventional serine/threonine phosphorylation enrichment methods (e.g., immobilized metal affinity chromatography, IMAC). Leveraging high-sensitivity mass spectrometry platforms and extensive phosphopeptide spectral libraries, this service enables systematic identification and analysis of phosphorylation sites on serine (S), threonine (T), and tyrosine (Y) residues. Its core strength lies in enhancing the detection depth of tyrosine phosphorylation sites (pY) to several times that of conventional methods, while significantly improving phosphorylation site coverage and functional annotation capabilities through multidimensional technology integration. This approach provides high-resolution dynamic phosphorylation maps to support drug target discovery, signaling pathway elucidation, and disease mechanism research.
Protein phosphorylation is one of the most widespread and critical post-translational modifications (PTMs) in biological systems. It mediates critical biological processes—including protein activity regulation, cellular signaling, metabolic pathways, and disease pathogenesis—through the reversible addition or removal of phosphate groups. Studies estimate that over 30% of proteins in the human proteome undergo phosphorylation, which plays pivotal roles in pathological mechanisms underlying cancer, neurodegenerative disorders, immune dysregulation, and other diseases.
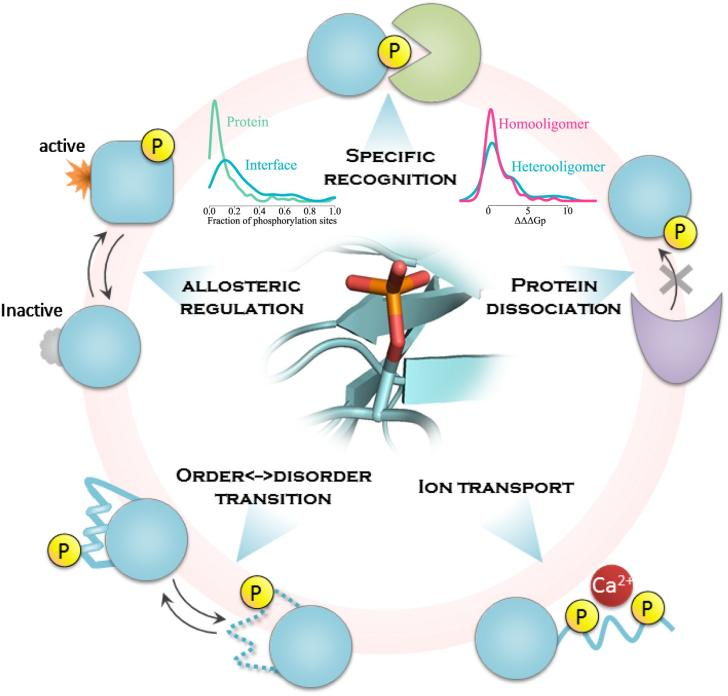
Nishi H, et al. Structure. 2011.
Figure1. Multiple Mechanisms of Protein Phosphorylation in Ccell Signaling Regulation
However, traditional phosphorylation studies are limited by technical challenges such as low sensitivity and insufficient coverage, making it difficult to comprehensively analyze phosphorylation sites and their dynamic changes in complex biological samples. With advancements in mass spectrometry and breakthroughs in omics methodologies, comprehensive phosphoproteomics has emerged as a critical tool for precisely decoding phosphorylation-mediated regulatory networks. Leveraging cutting-edge mass spectrometry platforms and proprietary enrichment technologies, MtoZ Biolabs introduces global phosphoproteomics service, designed to deliver high-precision, high-throughput phosphorylation analysis solutions for academic institutions, pharmaceutical companies, and clinical diagnostics.
Service at MtoZ Biolabs
As a mass spectrometry-driven leader in post-translational modification (PTM) proteomics, MtoZ Biolabs offers end-to-end global phosphoproteomics service—from sample preparation to data interpretation—powered by Orbitrap™ advanced mass spectrometry platforms, proprietary phosphorylation enrichment technologies, and a repository of millions of curated modification spectra. Our services include:
These capabilities empower researchers to address critical scientific challenges in signal transduction mechanism studies, drug target discovery, and precision medicine research.
Our Expertise
1. High-Depth Phosphorylation Site Identification:
Compatible with diverse sample types (cells, tissues, serum, etc.), enabling detection of 10,000+ phosphorylation sites in a single run.
2. Dynamic Quantitation Analysis:
Utilizes Label-free, TMT, or iTRAQ labeling technologies to profile phosphorylation changes across experimental conditions.
3. Ultra-High Sensitivity:
Supports single-cell-level phosphorylation detection for rare or limited samples.
4. Comprehensive Coverage:
Simultaneously profiles S/T/Y phosphorylation sites, with tyrosine (pY) detection depth several-fold higher than conventional methods.
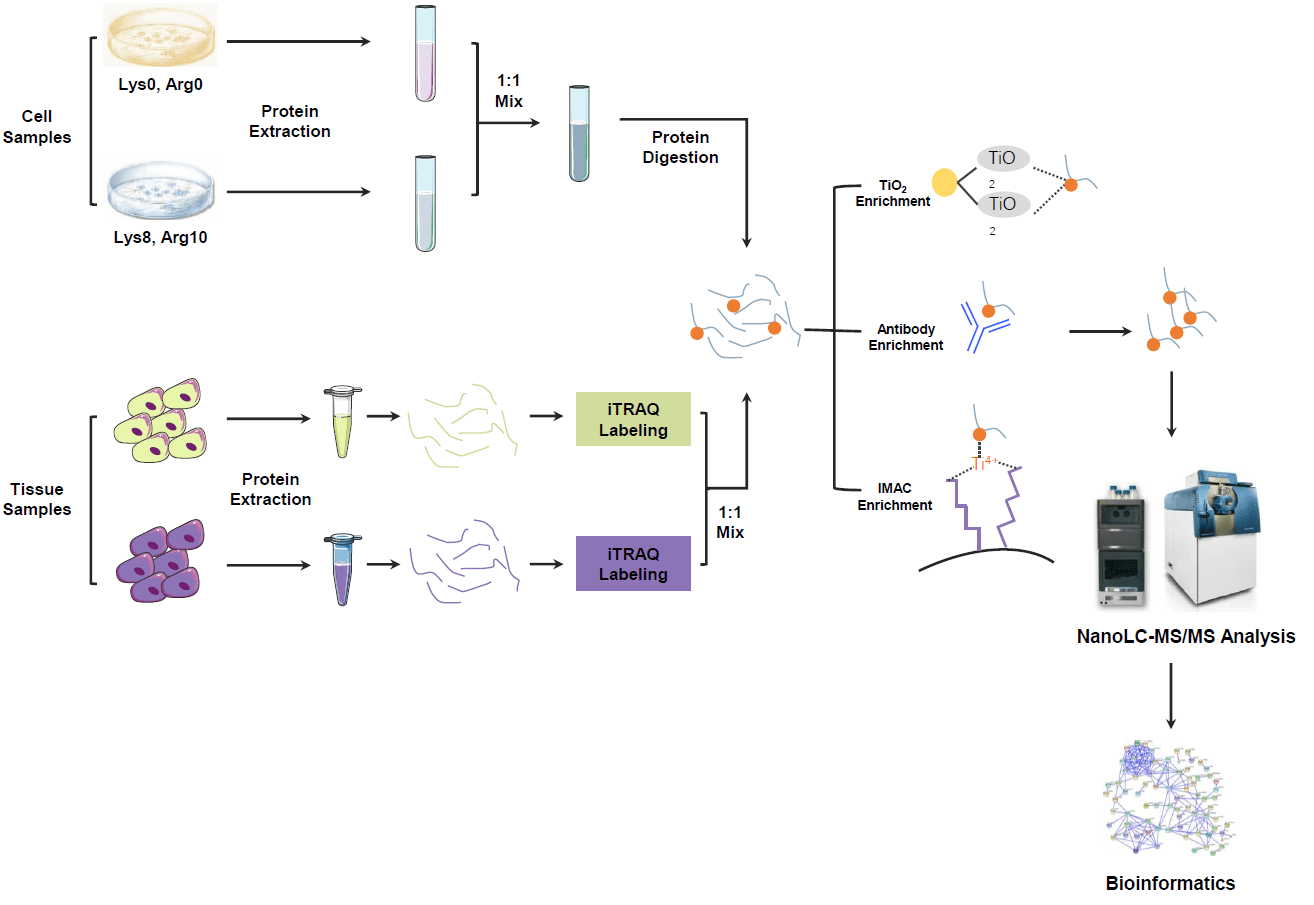
Figure2. Global Phosphoproteomics Service Workflow
Technical Principles
Global phosphoproteomics is grounded in the systematic capture and analysis of phosphorylation sites on serine (S), threonine (T), and tyrosine (Y) residues. Its core innovation lies in the integration of multidimensional enrichment technologies with high-sensitivity mass spectrometry platforms to achieve exhaustive coverage of phosphorylation sites. Traditional methods rely on immobilized metal affinity chromatography (IMAC) or metal oxide affinity chromatography (MOAC) to enrich serine/threonine (S/T) phosphorylated peptides. However, these approaches exhibit limited efficiency in capturing low-abundance tyrosine phosphorylation (pY). In contrast, the global phosphoproteomics service introduces tyrosine phosphorylation-specific antibody enrichment, coupled with IMAC/MOAC, to establish a dual-pathway enrichment system. This hybrid strategy dramatically enhances the detection depth of pY sites.
Following enrichment, phosphorylated peptides are analyzed using high-resolution mass spectrometry. Advanced techniques such as trapped ion mobility separation (TIMS) and data-independent acquisition (DIA) enable ultra-sensitive detection of low-input samples. Simultaneously, robust phosphorylation spectral libraries ensure precise site localization and quantitation.
Applications
1. Drug Target Discovery & Kinase Inhibitor Development
Precisely mapping kinase-substrate regulatory networks to accelerate target screening and mechanistic studies for small-molecule inhibitors (e.g., osimertinib, lapatinib).
2. Disease Mechanism & Biomarker Discovery
(1) Uncover dynamic phosphorylation imbalances in diabetes and neurodegenerative diseases using comprehensive phosphoproteomics. For example, elucidate the pivotal role of the GSK3-PDX1 axis in pancreatic β-cell dysfunction.
(2)Identify tau protein phosphorylation biomarkers in cerebrospinal fluid for early Alzheimer’s disease diagnosis.
Case Study
Phosphorylation is a critical post-translational modification that regulates skeletal muscle metabolism and exercise adaptation. However, the global phosphorylation network in human skeletal muscle during exercise remains poorly characterized, particularly the relationships between exercise-induced kinases and their substrates. This study employed global phosphoproteomic analysis, integrating liquid chromatography-tandem mass spectrometry (LC-MS/MS) with phosphopeptide enrichment methods, to systematically profile pre- and post-exercise phosphorylation changes in human skeletal muscle. A kinase-substrate network regulated by exercise was constructed.
Key Findings:
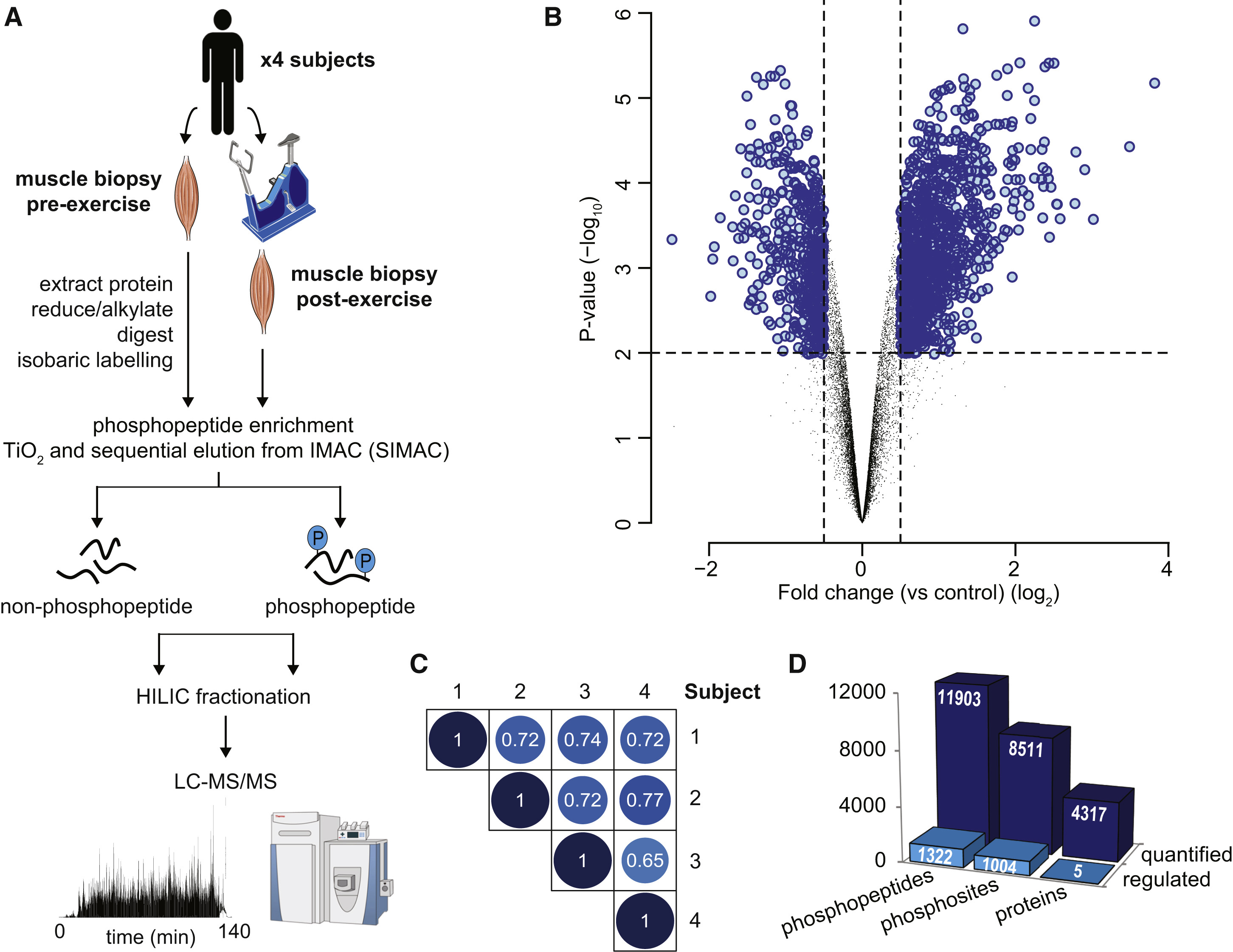
Hoffman, N. J. et al. Cell Metab. 2015.
Figure3. Acute Exercise-Regulated Phosphoproteome in Human Skeletal Muscle
This work establishes the first exercise-associated global phosphorylation map in human skeletal muscle, clarifying AMPK’s critical role and its substrate interaction network during exercise. These findings provide a foundational dataset and novel research directions for understanding exercise adaptation mechanisms, metabolic regulation, and therapeutic strategies for exercise-related disorders.
FAQ
Q1: Why do conventional phosphoproteomics and comprehensive phosphoproteomics yield inconsistent results for pY site detection? How should I choose between them?
A: Comprehensive phosphoproteomics employs tyrosine phosphorylation-specific antibody enrichment, significantly enhancing pY detection sensitivity and specificity. For example, traditional IMAC enrichment typically captures only 1-3% pY sites, whereas the comprehensive approach achieves over 20% pY coverage. If conflicting trends arise, prioritize results from the comprehensive technology, as it minimizes nonspecific binding interference and leverages large-scale spectral libraries to reduce false positives.
Q2: Are phosphorylation functional analyses based on individual modification sites or the entire protein?
A: Existing functional databases (e.g., KEGG, GO) annotate proteins holistically. However, comprehensive phosphoproteomics enables site-specific functional insights through Kinase Substrate Enrichment Analysis (KSEA) and motif prediction, pinpointing upstream kinases and regulatory modules. For instance, elevated EGFR kinase activity may correlate with MAPK pathway activation, but site-specific functional validation (e.g., via mutagenesis or multi-omics integration) is essential to confirm causality.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

