





Discovery Proteomics Service
Discovery proteomics is an unbiased proteomics research approach aimed at the comprehensive identification and quantification of all detectable proteins in a biological system. Its core principle relies on liquid chromatography-mass spectrometry (LC-MS/MS), utilizing either data-dependent acquisition (DDA) or data-independent acquisition (DIA) strategies to perform high-throughput screening and in-depth analysis of proteins in complex biological samples. Unlike targeted proteomics, discovery proteomics does not predefine target proteins; instead, it systematically analyzes all proteins in a sample to identify differentially expressed proteins, recognize key biomarkers, and explore protein interaction networks and regulatory mechanisms.
Discovery proteomics service is widely applied in disease research, biomarker discovery, drug target identification, immunology, metabolic regulation, precision medicine, agriculture, and food science. For example, in cancer research, this technology can be used to screen tumor-associated proteins, revealing mechanisms of tumor initiation, progression, and drug resistance. In neurodegenerative disease research, it aids in identifying potential pathogenic proteins and regulatory pathways involved in conditions such as Alzheimer’s and Parkinson’s disease. Additionally, discovery proteomics is valuable for investigating drug action mechanisms, studying post-translational modifications (PTMs), constructing protein interaction networks, and ensuring the quality control of biopharmaceuticals, providing critical data support for life sciences research and precision medicine.
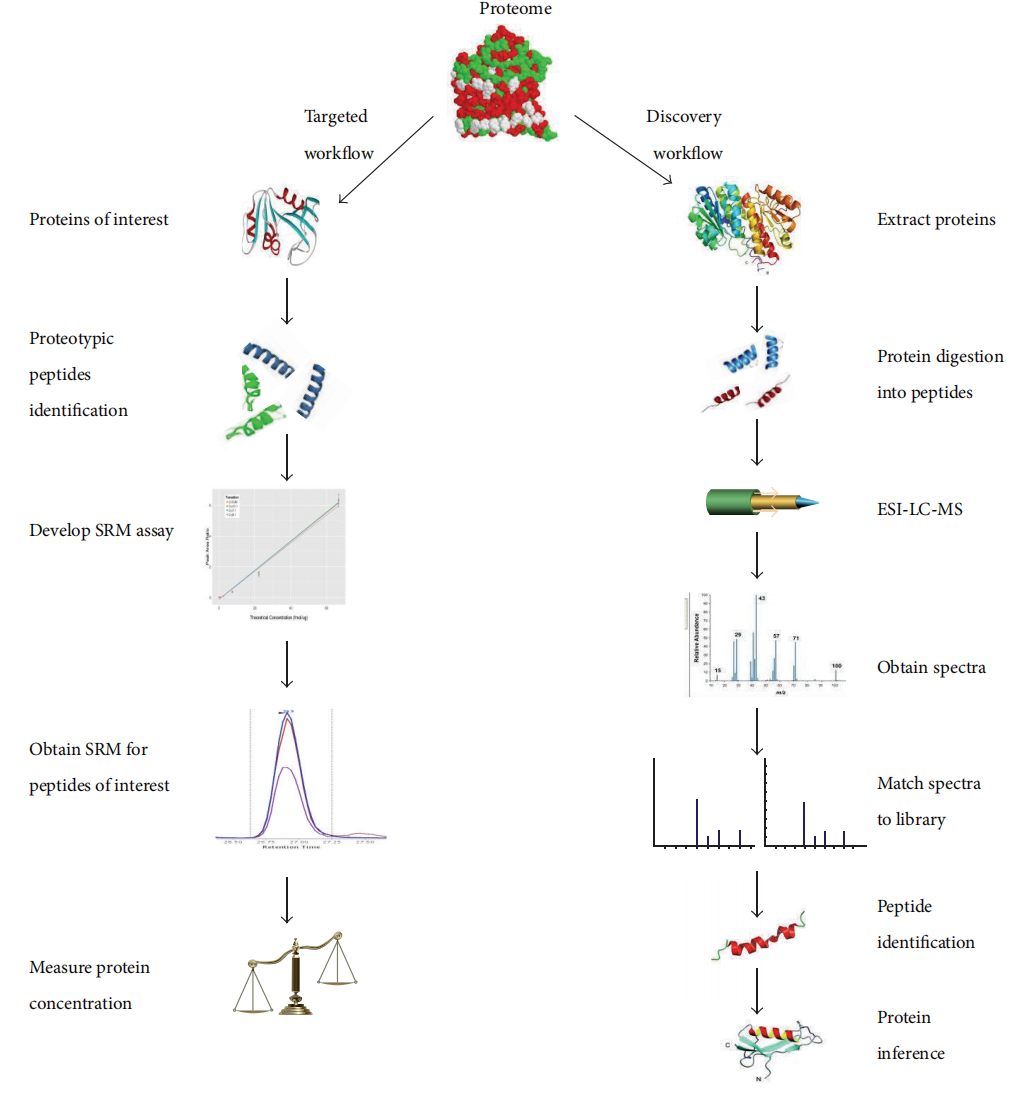
Mesri, M. Adv Med. 2014.
Figure 1. Comparision Between Targeted Proteomics and Discovery Proteomics.
Service at MtoZ Biolabs
Utilizing high-resolution liquid chromatography-mass spectrometry (LC-MS/MS) technology, MtoZ Biolabs offers discovery proteomics service that ensure high-sensitivity and high-resolution data acquisition. Our services encompass comprehensive sample preparation, protein extraction, enzymatic digestion, liquid chromatography separation, DDA/DIA mass spectrometry detection, and bioinformatics analysis, enabling thorough protein identification and quantification within biological samples. We guarantee high accuracy and reproducibility, providing deep proteome coverage. Additionally, we offer quantitative protein expression analysis, post-translational modification (PTM) identification, protein interaction analysis, and large-scale phosphoproteomics studies, supporting fundamental research, disease mechanism exploration, and drug target discovery.
Service Advantages
1. High-throughput Analysis and Comprehensive Coverage
MtoZ Biolabs' discovery proteomics service utilizes advanced mass spectrometry technology to perform high-throughput analysis of complex biological samples, identifying and quantifying thousands of proteins. This unbiased approach avoids the limitations of focusing only on known proteins, providing a comprehensive view of the proteome and helping researchers explore previously unknown proteins and potential biomarkers.
2. Accurate Quantification and High Reproducibility
By integrating various quantification strategies such as Label-Free, TMT, DIA, and PRM, we ensure high accuracy and sensitivity in protein expression data. Our approach supports large-scale sample analysis, meeting diverse research needs with high reproducibility.
3. Comprehensive Bioinformatics Analysis
MtoZ Biolabs not only provides raw mass spectrometry data but also offers in-depth data analysis and interpretation. Our services include differential protein screening, GO/KEGG pathway enrichment analysis, protein-protein interaction (PPI) network construction, and multi-omics integration, facilitating disease research, drug target screening, and precision medicine applications.
4. Customized Experimental Design
We offer one-on-one scientific consultation tailored to specific research requirements, allowing researchers to select the most suitable experimental strategy. Our flexible approach ensures the efficient achievement of research objectives.
Applications
1. Disease Research and Biomarker Discovery
Through comprehensive protein expression analysis, we identify disease-associated proteins, facilitating early diagnosis and precision medicine for cancer, neurodegenerative diseases, cardiovascular disorders, and more.
2. Drug Development and Target Screening
Discovery proteomics service supports new drug screening, drug mechanism research, target identification, and drug response evaluation, optimizing drug development and personalized treatment strategies.
3. Immunology and Tumor Microenvironment Research
By analyzing immune-related protein expression patterns, discovery proteomics explores inflammatory responses, tumor immune microenvironments, and cytokine regulation, contributing to immunotherapy research.
4. Post-Translational Modification (PTM) Analysis
Discovery proteomics service can detect post-translational modifications (PTMs) such as phosphorylation, acetylation, and ubiquitination, enabling the study of protein functional regulation mechanisms and revealing the dynamic modulation of cellular signaling pathways.
5. Biopharmaceutical Development and Quality Control
In the development of biologics, including antibody drugs, vaccines, and protein therapeutics, discovery proteomics monitors protein expression, purity, and modifications to ensure product quality and stability.
6. Agriculture and Food Science
Discovery proteomics service is applied in crop stress resistance research, livestock health monitoring, food safety testing, and functional food component identification, providing precise protein data for agricultural biotechnology and food science.
Case Study
1. Discovery Proteomics Reveals Potential Protein Signature Associated with Malignant Phenotype Acquisition in Pleomorphic Adenoma
The study aims to investigate the molecular mechanisms underlying the malignant transformation of Pleomorphic Adenoma (PA) into Carcinoma Ex-Pleomorphic Adenoma (CXPA) using discovery proteomics and to identify potential protein signatures associated with malignant phenotype acquisition. Tissue samples from PA and CXPA patients were analyzed using LC-MS/MS for comprehensive proteomic profiling, followed by bioinformatics analysis to identify differentially expressed proteins (DEPs) and their functional enrichment in biological pathways. The results revealed significant protein expression changes in CXPA compared to PA, particularly in processes related to extracellular matrix remodeling, epithelial-mesenchymal transition (EMT), proliferative signaling pathways, and inflammatory regulation. Notably, members of the MMP protein family, integrin-associated proteins, and Wnt signaling regulators were markedly upregulated in CXPA, suggesting their critical roles in malignant transformation. Furthermore, protein interaction network analysis indicated that these DEPs might contribute to malignant phenotype acquisition by modulating the tumor microenvironment. The study concludes that discovery proteomics can effectively identify potential biomarkers for PA-to-CXPA progression and provide valuable insights into the molecular mechanisms driving this transformation, supporting the development of early diagnostic strategies.
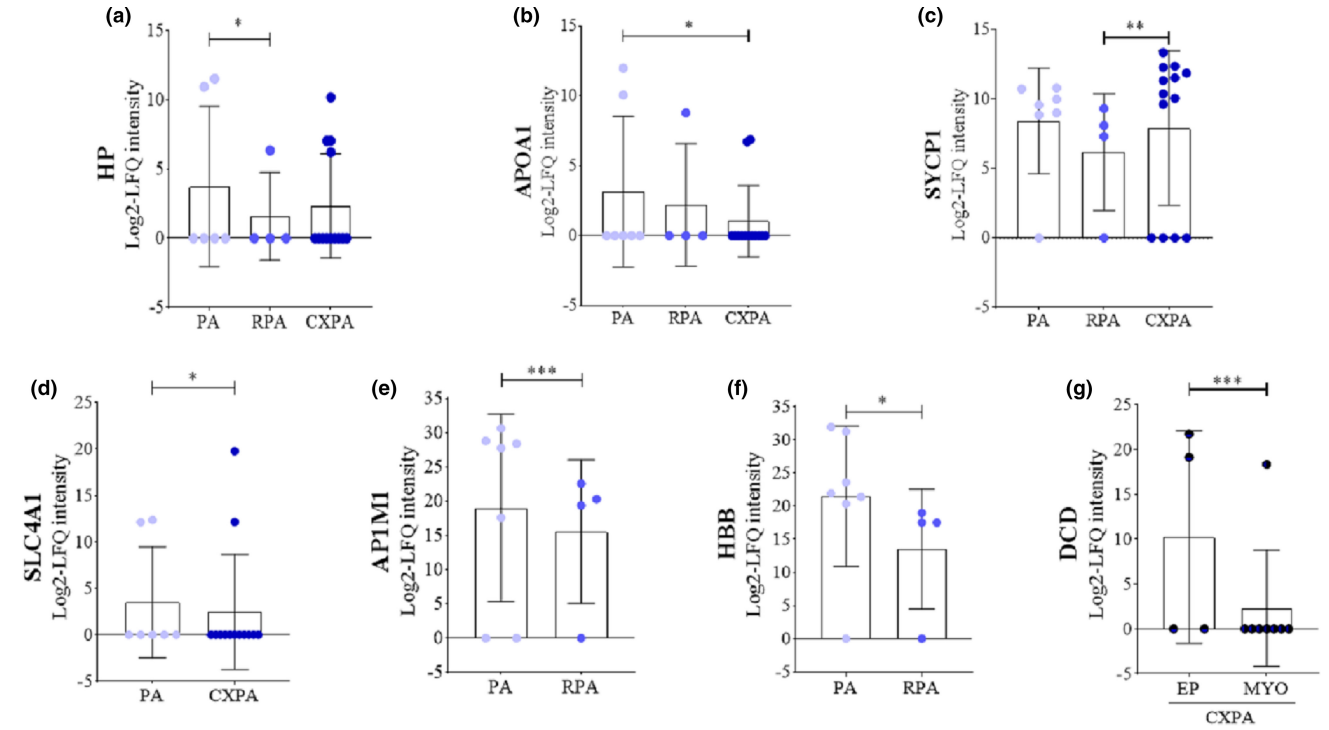
Lima-Souza, R A D. et al. Oral Dis. 2023.
Figure 2. Log2-LFQ Intensity of the Candidate Proteins for Protein Signature.
FAQ
Q: How can the quantitative accuracy and sensitivity for low-abundance proteins be improved in discovery proteomics analysis, while reducing biases and errors in data processing, to more accurately reflect the true protein expression in biological samples?
A: First, optimizing sample preparation is essential, as improving sample extraction efficiency and minimizing protein degradation can enhance the accuracy of quantitative results. Secondly, the sensitivity of mass spectrometry analysis is key. Using high-resolution mass spectrometers and high-sensitivity ionization techniques, such as electrospray ionization (ESI) or MALDI, can significantly improve the detection of low-abundance proteins. Furthermore, optimizing data analysis algorithms is also critical. Employing more precise quantification methods, such as labeling approaches (SILAC, TMT) or label-free quantification, combined with complex statistical methods, can effectively reduce data bias and improve analysis accuracy. Finally, standardization and validation across laboratories are essential. Through multiple experimental validations and repeat analyses with various samples, the reliability of the data can be further confirmed, ensuring that discovery proteomics results reflect the true biological state. The integration of these technologies and strategies can help overcome the challenges of quantifying low-abundance proteins.
Deliverables
1. Comprehensive Experimental Details
2. Materials, Instruments, and Methods
3. Relevant Liquid Chromatography and Mass Spectrometry Parameters
4. The Detailed Information of Proteomics
5. Mass Spectrometry Image
6. Raw Data Files
MtoZ Biolabs, an integrated Chromatography and Mass Spectrometry (MS) Services Provider, provides advanced proteomics, metabolomics, and biopharmaceutical analysis services to researchers in biochemistry, biotechnology, and biopharmaceutical fields. Our ultimate aim is to provide more rapid, high-throughput, and cost-effective analysis, with exceptional data quality and minimal sample consumption. Free project evaluation, welcome to learn more details!
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

