





De Novo Sequencing vs Reference-Based Sequencing: What’s the Difference?
Different research objectives and sample types demand distinct sequencing approaches. This is especially true in applications such as proteomics or antibody analysis, where researchers often face confusion when choosing between De Novo sequencing and reference-based sequencing: What fundamentally differentiates the two? Under what circumstances is De Novo essential? And why has it become a pivotal technology in current protein structure analysis? This article aims to clarify the distinctions and selection criteria for these two sequencing strategies across three dimensions: underlying principles, application scenarios, and comparative advantages.
1. Principle Differences: Reference-Based Sequencing vs De Novo Sequencing
Reference-based sequencing, also referred to as database-dependent mass spectrometry analysis, involves matching the peptide fragment spectra obtained from mass spectrometry with theoretical spectra derived from known protein databases such as Uniprot or RefSeq. Since this method relies on established protein information, it offers high matching efficiency, reduced computational burden, and relatively high accuracy. It is particularly suited to model organisms or research subjects with well-annotated reference sequences.
In contrast, De Novo sequencing infers amino acid sequences directly from MS/MS spectral data without relying on any existing database. Advanced algorithms identify fragment ions (typically b/y ion series), deduce the sequences of individual peptides, and further assemble them into the full primary structure of proteins. Even in the absence of database records, De Novo can reconstruct the actual sequence, making it a truly data-driven approach suitable for studying unknown or highly variable proteins.
2. Application Scenarios: When Is Reference-Based Sequencing Sufficient? When Is De Novo Necessary?
For model organisms such as humans, mice, fruit flies, and yeast, comprehensive genomic and proteomic annotations exist, enabling reference-based sequencing to cover over 90% of known proteins. This approach is highly efficient and robust for routine proteomic tasks, including expression profiling, phosphorylation site identification, and quantitative protein screening.
However, in several critical scenarios, reference-based strategies become inadequate, necessitating the use of De Novo sequencing:
(1) Reconstruction of unknown antibody structures: including those derived from animal immunization, clinical samples, or patent antibodies lacking sequence entries in public databases.
(2) Non-model organism studies: such as investigations into traditional medicines, microorganisms, or marine species with unannotated or incomplete proteomic data.
(3) Detection of mutated proteins or neoantigens: such as novel proteins expressed in cancer neoantigens or viral variants.
(4) Post-translational modifications interfering with database matching: including glycosylation or deamidation events that compromise standard database searches.
(5) Verification of expression consistency across systems: for example, assessing whether an antibody exhibits minor structural differences when produced in different expression platforms.
In these contexts, De Novo sequencing not only retrieves the authentic protein sequences but also reveals structural variants, modification isomers, and splice forms that are typically missed by database-based methods. As such, it serves as a fundamental technique for deep structural characterization and precision drug development.
3. Comparison of Advantages and Disadvantages: The Choice Is Not Just About "Accuracy"
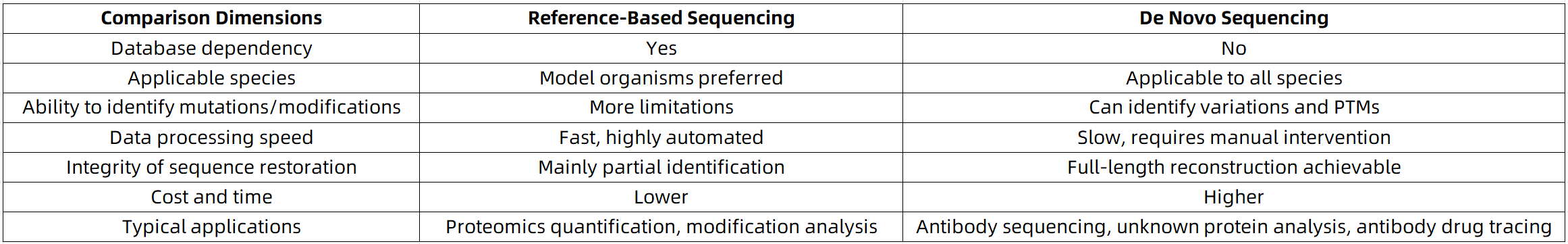
MtoZ Biolabs' Dual-Strategy Protein Sequencing Solution
At MtoZ Biolabs, we offer not only conventional proteomics services based on reference databases, but also advanced full-length De Novo sequencing at the antibody level. We have extensive project experience in high-complexity structural characterization scenarios, including: reconstruction of antibody light and heavy chain sequences, identification of dominant clones within polyclonal antibodies, structural elucidation of vaccine-induced antibodies, characterization of modified protein isoforms, and consistency assessment of biosimilar antibodies. Our approach integrates multi-enzyme digestion strategies, the Orbitrap high-resolution mass spectrometry platform, proprietary De Novo sequence assembly algorithms, and manual curation supported by structure-based modeling. This workflow enables us to deliver complete protein sequences with high coverage, expressibility, and experimental verifiability.
If your research focuses on differential protein expression or functional annotation, reference-based mass spectrometry remains a highly efficient and robust option. However, when facing a novel protein absent from current databases, or when a peptide ID alone is insufficient and a reproducible, expressible full-length sequence is required, De Novo sequencing becomes the method of choice. MtoZ Biolabs is committed to empowering researchers and biopharmaceutical developers to choose the most appropriate sequencing strategy and obtain accurate, complete, and functionally meaningful protein structural information. Whether you are conducting basic research, developing therapeutic antibodies, or performing biosimilarity assessments, we provide comprehensive technical support and reliable data delivery tailored to your needs.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

