





Cryo-EM Single Particle Analysis Service
The cryo-EM single-particle analysis (SPA) involves preparing numerous molecular samples with identical structures. These are dispersed and vitrified for random projection imaging, followed by computational angle determination and the alignment of particles with matching angles to emphasize distinctive, interpretable features. Assuming uniformity in molecular structure, the technique involves statistical analysis of multiple images and image processing to enhance the signal-to-noise ratio. After confirming the spatial relationships among the two-dimensional (2D) images, three-dimensional (3D) reconstruction is performed to deduce the biomolecule's structure.
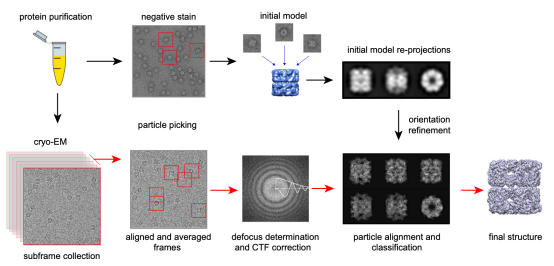
Figure 1. The Process of Cryo-EM SPA Technique [3]
The single-particle cryo-EM workflow is as follows:
1. Sample Purification
Upon selection of the target protein for cryo-EM analysis, plasmid construction and protein expression are carried out via prokaryotic or eukaryotic methods. Post purification, efforts to replicate the native environment of the macromolecule are made to obtain accurate structural data. This involves tuning the pH and adding salts, glycerol, detergents, and other additives to simulate the native conditions within the solution.
Typical cryo-EM buffer conditions align with cellular environments, usually maintaining pH between 7 and 8.5 with suitable salt concentrations. Nevertheless, the optimal pH for many macromolecules ranges widely from 4.5 to 9. Using less-than-ideal buffer conditions can lead to production challenges, thermodynamic instability, and increased aggregation and heterogeneity. To mimic cellular crowding, additives like glycerol or sucrose are utilized, but their high concentrations may compromise high-resolution imaging by negatively affecting vitrification and reducing contrast.
During sample film formation, the high surface-to-volume ratio can challenge protein stability, possibly leading to denaturation or aggregation. Protein samples are categorized into types such as membrane proteins and larger macromolecular protein complexes. Cytosolic, or soluble, proteins have a tendency to remain stable in thin films owing to the hydrophilic amino acids on their external structures. However, membrane proteins often require extra solvents for stability, with their hydrophobic amino acids embedded within the lipid bilayer, making them susceptible to air-water interface effects. Agents like detergents, amphiphiles, nanodiscs, and styrene-maleic acid copolymers are employed to stabilize these proteins by mimicking the lipid bilayer, enhancing the affinity around the hydrophobic regions. Additional strategies for handling delicate proteins include blocking the air-water interface during purification and adding certain agents before deposition onto grids to isolate them from the interface. Larger structures are preserved through chemical cross-linking to maintain integrity.
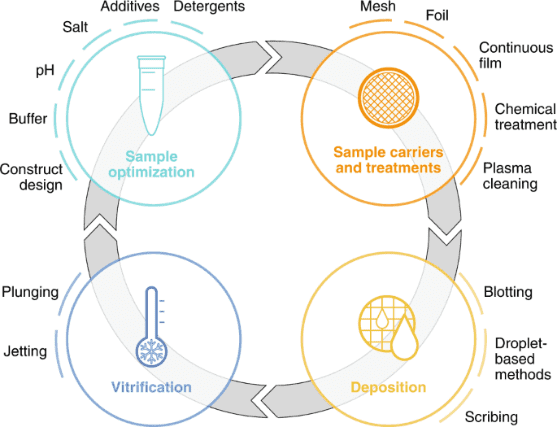
Figure 2. Cryo-EM Sample Preparation Workflow [4]
2. Sample Restaining Screening
After purification, it is essential to evaluate the quality of the purified sample to determine its suitability for further EM analysis. Even complete complexes may consist of mixtures made up of different subcomplexes, and homologous complexes might exhibit multiple conformations. Negative staining is a method used to observe particulate substances or biomolecules within a sample. Its fundamental principle involves adsorbing protein particles onto a grid coated with a hydrophilic membrane. The background near the protein particles is then stained with heavy metal salts, making the protein particles visible under the electron microscope because they are not stained by these salts. Common heavy metal salts used for negative staining include ammonium molybdate, methylamine tungsten, methylamine vanadium, sodium/potassium phosphotungstate, sodium silicotungstic, uranyl acetate, and uranyl formate.
Negative stain EM is a convenient method for assessing the quality of purified biological samples under a microscope. This screening is designed to qualitatively evaluate the uniformity of particle composition and conformation, and it can only be performed under microscopic observation. This serves as a rapid prescreening method prior to cryo-EM, facilitating the evaluation of the sample's uniformity and its buffer components.
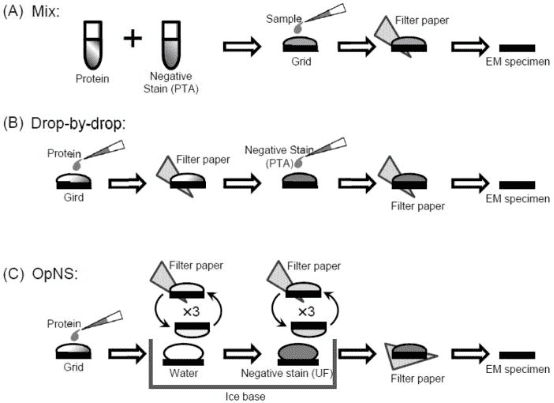
Figure 3. Common Negative Dyeing Processes [5]
While negative staining provides general information about the molecules, it does not reveal high-resolution details. Despite the benefits of this screening method, challenges such as molecular dissociation at the air-water interface or concentration-dependent aggregation may still occur when preparing samples in a vitrified state. This can affect the quality and interpretability of the images produced by EM.
3. Sample Preparation
The primary process for sample preparation involves depositing high-purity, high-concentration protein solutions onto a specially designed grid consisting of a thin, perforated amorphous carbon film supported by a metal frame. This setup facilitates the formation of a uniform thin water film across the holes due to surface tension. After removing excess solution, the grid with the protein film is rapidly plunged into liquid ethane for vitrification, securing the proteins within a vitreous ice layer.
Sample carriers are historically circular EM grids of 3 mm in diameter that consist of at least two components: a mesh base and foil. The mesh base is made of metal to attain mechanical stability, conduction of the electron beam and heat dissipation. Commercially available meshes range from 200-400 grid bars per inch and are made of copper, gold, or nickel. The choice of grid geometry and the reduction of pore size minimize thermal deformation. To prevent adherence of sample particles to the substrate, continuous thin films made of carbon or graphene oxide are employed, which also increases particle density per pore. These films are often chemically treated to enhance binding affinity. To ensure effective sample deposition, the carrier must be wettable, with plasma treatment commonly used to clean and temporarily enhance the grid's wettability. Sample loading techniques include blotting, droplet-based and scribing methods.
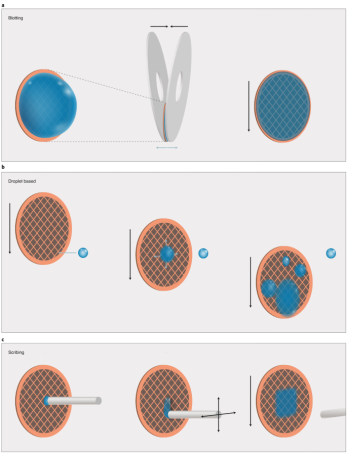
Figure 4. SPA Cryo-EM Sample Film Preparation Methods [4]
To provide useful information on the structure of target biomolecules as a function of time in cryo-EM experiments, it is essential to initiate and appropriately control the timing of the reaction. Various sample preparation methods have utilized different techniques for initiating and controlling reaction times: the "spraying-mixing" method employs the principle of passive diffusion, spraying two reactants in sequence onto the same grid, then flash-freezing within less than 100 milliseconds; the "mixing-spraying" method mixes the sample in a microfluidic device before loading it onto the grid; and specific wavelengths of light can also be used to trigger reactions.
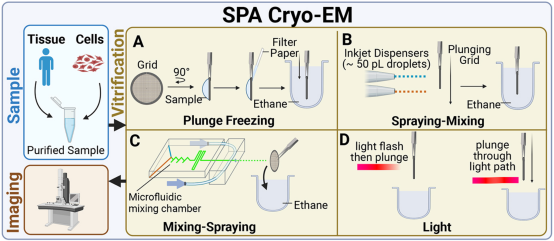
Figure 5. Time-Resolved SPA Cryo-EM Sample Preparation Methods [5]
After preparing the ultra-thin layer of sample solution on the copper grid, the grid must be quickly immersed into liquid ethane to rapidly freeze the sample. Liquid ethane has a very high thermal conductivity, which can quickly reduce the temperature of the sample solution to the vitreous state, thus achieving the preparation of vitreous state samples. Therefore, after the vitreous state sample is prepared, the copper grid can be transferred from the liquid ethane to liquid nitrogen for storage. The vitreous state sample remains stable at the low temperatures of liquid nitrogen, which facilitates long-term storage and transportation of the sample.
4. EM Image Acquisition
Single-particle data collection entails gathering thousands of micrographs from samples, selecting those with optimal particle density and vitreous ice thickness for producing the best images. Optimal parameters such as defocus values, magnification levels, and electron doses are set, and extensive images of these sample areas are recorded, with discrete molecular projections framed manually or via semi-automatic programs.
Although largely automated, the collection process can still be improved. The most unproductive step during data acquisition is the 15–30-s wait for the sample stage mechanical drift to settle after a stage move. To eliminate such pauses, the acquisition area can be moved optically instead of mechanically. Optical shifting of the beam using beam deflectors is fast but introduces off-axis optical aberrations, which could limit the achievable resolution. New strategies in optics and image processing have been developed to address these issues. The first one involves compensating the offaxial aberrations through 'on-the-fly' optical tuning (beam-tilt compensation); the second uses software demodulation of the aberrations during image processing. In testing, both methods produced similar outcomes and in practice the software demodulation approach is much easier to use. For example, in Figure 8, light blue circles represent thin free-standing ice regions where images are collected supported on a holey thin film (grey background). Traditionally, samples are moved mechanically from one aperture (A) to the next (B), requiring significant mechanical drift relaxation time before capturing each image. The novel beam-shift method positions the microscope stage at a central aperture (A) and optically shifts the imaging area to multiple adjacent apertures (C), enhancing data collection speed by tenfold.
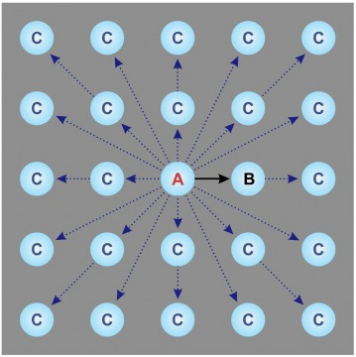
Figure 6. An Optimized High-Throughput SPA Data Collection Scheme [6]
Real-time monitoring and evaluation of data collected have become standard features on most high-end cryo-EM instruments. Initially, this functionality was employed to monitor the technical quality of the data. Parameters of each micrograph, such as resolution cut-off, amount of defocus, astigmatism, and drift rate, provide an overview of data collection performance. Currently, the biochemical and structural aspects of the data are also under surveillance. For instance, immediate automatic particle picking and 2D classification yield insights into the purity, homogeneity, orientation distribution, and conformational and assembly states of macromolecules within the sample.
5. 3D Reconstruction
The cryo-EM image processing workflow has been standardized over the past decade, beginning with computational corrections for beam-induced specimen motion in collected images and frame filtering to account for cumulative radiation damage (dose-weighting). Corrected micrographs are processed using template-based or Gaussian algorithms for identifying biological specimens, followed by 2D rotational registration, classification, and averaging of particle images to screen out low-quality particles preliminarily. Selected particle subsets then undergo 3D registration and iterative classification using projection matching to assign angles and separate conformational and compositional heterogeneities. These states' angle assignments are refined to enhance 3D reconstruction resolution. Further quality and resolution enhancements are achieved through refined particle motion correction and aberration adjustment, restoring high spatial frequency information.
Developments in image processing software are crucial for Cryo-EM single-particle reconstruction, with ongoing advances in efficient contrast transfer function (CTF) fitting, maximum likelihood estimation, stochastic gradient descent, multibody refinement, Bayesian polishing, and CTF refinement significantly impacting result quality. Prominent EM analysis software systems include SPIDER, EMAN2, FREALIGN, SPARX, and RELION.
Analysis Workflow
1. Experimental Procedure Determination Based on Requirements
2. Plasmid Construction and Protein Purification
3. Sample Counterstain Screening and Optimization
4. Sample Preparation
5. Data Collection
6. 3D Reconstruction
Service Advantages
1. Customizable Experimental Methods Based on Requirements
2. Multi-Gradient Design for Optimal Condition Exploration and Sample Optimization Guidance
3. Automated Sample Preparation Platform for Precise Reaction Condition Control
4. High-Resolution Cryo-EM Image Acquisition
5. Diverse Data Analysis Techniques
Sample Results
1. Single-Particle Cryo-EM Reconstruction of 52 kDa Streptavidin at 3.2 Å Resolution
The rapid advancements in single-particle cryo-EM have made it more feasible to obtain the 3D structure of well-behaved macromolecules with a molecular weight higher than 300 kDa at ~3 Å resolution. Yet, achieving high-resolution for molecules under 200 kDa remains challenging. This study utilized the Cs-corrector-VPP-coupled cryo-EM to investigate a 52 kDa streptavidin (SA) protein supported on graphene and embedded in vitreous ice, achieving near-atomic resolution structures of both the apo-SA and biotin-bound SA. This method has shown potential for resolving structures of molecules as small as 39 kDa.
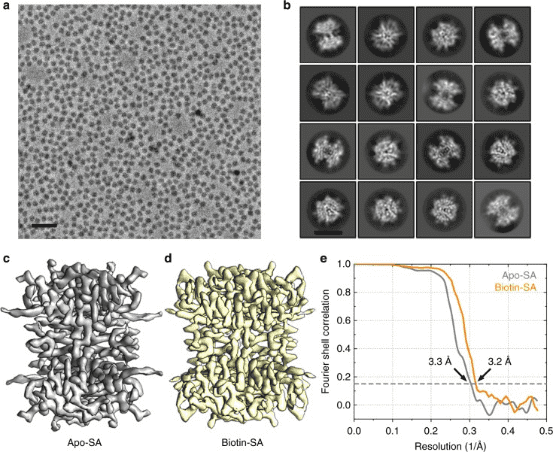
Figure 7. SPA Cryo-EM Resolved Structure of SA [7]
2. Atomic Resolution Analysis of Human Membrane Protein β3 GABA by Single-Particle Cryo-EM
Recent breakthroughs in electron detection and image processing have increasingly detailed the structures of proteins obtained by cryo-EM SPA. However, obtaining sufficient resolution for cryo-EM reconstruction to observe individual atoms within proteins remains challenging. Research has shown that with a new electron source, energy filter, and camera to obtain a 1.7 Å resolution cryo-EM reconstruction for a human membrane protein—the β3 GABAA receptor homopentamer3—was achieved. This spectrum allows for a detailed understanding of small molecule coordination, visualization of solvent molecules, alternative conformations for multiple amino acids, and unambiguous building of ordered acidic side chains and glycans. Applying this strategy to mouse apoferritin achieved a reconstruction at 1.22 Å resolution, providing a genuine atomic-resolution view of a protein molecule with single-particle cryo-EM for the first time. The described technological advancements, combined with further methods to accelerate data collection and improve sample quality, provide a pathway for routine application of cryo-EM in high-throughput screening of small molecule modulators and structure-based drug discovery.
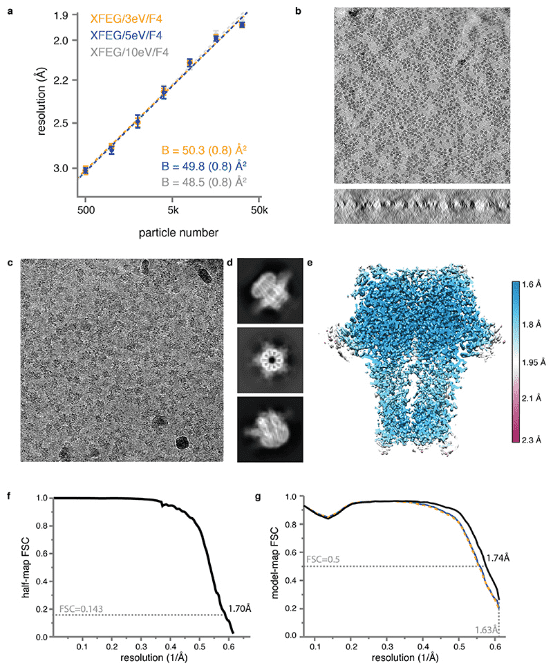
Figure 8. GABAA Receptor Structure at 1.7 Å Resolution [8]
3. Structural Basis of Oligomerization of Helicobacter pylori VacA Toxin Revealed by Cryo-EM
Helicobacter pylori colonizes the human stomach and contributes to the development of gastric cancer and peptic ulcer disease. Helicobacter pylori secretes a pore-forming toxin known as vacuolating cytotoxin A (VacA), which consists of two domains (p33 and p55) and assembles into oligomeric structures. Using single-particle cryo-EM, low-resolution structures of VacA dodecamers and heptamers, as well as a 3.8 Å structure of a VacA hexamer, were determined. These analyses show that VacA p88 is primarily composed of a right-handed β-helix extending from the p55 domain to the p33 domain. The regions of p33 and p55 involved in hexamer assembly were mapped, simulating how interactions between protomers support heptamer formation, and identify surfaces of VacA that likely contact membrane. This work provides structural insights into the oligomerization process of VacA and identifies regions of the VacA protomer expected to contact the host cell surface during the channel formation process.
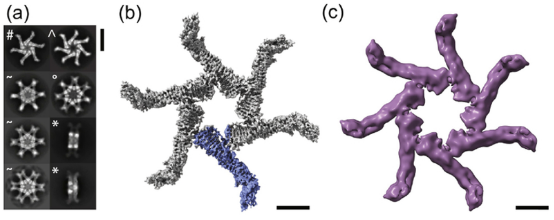
Figure 9. Cryo-EM Analysis of VacA s1m1 Oligomers [9]
Sample Submission Requirements
1. Comprehensive Experimental Steps
2. Specifications of Relevant Instrumentation
3. Original Experimental Data
4. Data Analysis Report
Applications
1. Improvement of Experimental Process Methods
Cryo-EM is widely used for structural biology research and has seen extensive development in recent years. However, the vitrification process of its samples can lead to severe particle aggregation and/or denaturation, thus being a major limiting factor. This effect is believed to be due to particles tending to adhere to the "deadly" air-water interface during vitrification. Therefore, a study reported a protein PEGylation method that effectively protects particles from such issues during the vitrification process. This method alleviates the laborious process of fine-tuning the vitrification conditions, allowing for analysis of samples that would otherwise be discarded.
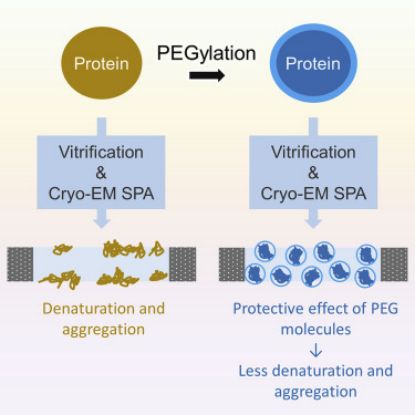
Figure 10. Improving Particle Quality in Cryo-EM Analysis Using the PEGylation Method [10]
2. Optimizing Experimental Processes through Multi-Technological Integration
Recent advancements allow single-particle cryo-EM to detail the structures of vast protein assemblies at high resolutions, providing unprecedented functional insights. However, despite its extraordinary capabilities, cryo-EM is still time-consuming and resource-intensive. Therefore, a method for quickly assessing and optimizing sample quality before undertaking extensive cryo-EM analysis is highly beneficial. To address this, research has developed a native mass spectrometry (nMS) platform that offers rapid feedback on sample quality and significantly simplifies biochemical screening. Because nMS can precisely analyze the mass of protein complexes, it is ideally suited for routine evaluation of the composition, integrity, and uniformity of samples prior to their plunge-freezing on EM grids.
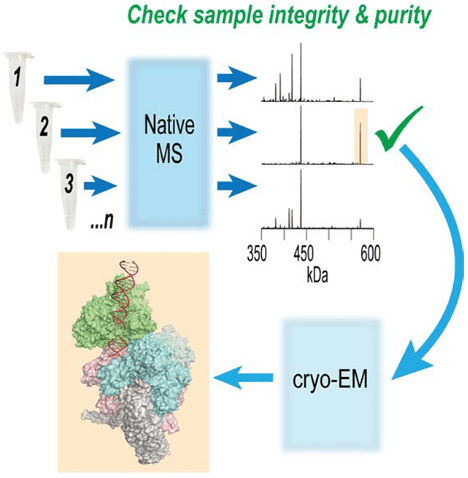
Figure 11. Optimizing Cryo-EM Sample Preparation with nMS Screening [11]
3. AI-Optimized Data Processing Workflows
Cryo-EM is widely used to determine the 3D structures of macromolecules. Due to the diversity of particle shapes and the extremely low signal-to-noise ratio of micrographs, picking particles from 2D micrographs remains a challenging early step. Because of these issues, extensive human intervention is usually necessary to generate a set of high-quality particles for input into downstream structural determination steps. A study introduced a fully automated method (DeepCryoPicker) for single-particle picking based on deep learning. It first uses automatic unsupervised learning to generate a particle training dataset. Then, it trains a deep neural network to automatically classify particles. Results show that DeepCryoPicker has advantages over semi-automated methods such as DeepEM, DeepPicker, and RELION, with the significant advantage of not requiring human intervention.

Figure 12. Overview of the DeepCryoPicker Program [12]
FAQ
Q1: What are the advantages and disadvantages of Cryo-EM SPA?
Compared to X-ray diffraction, the greatest advantage of the single-particle method is that it eliminates the time-consuming and labor-intensive protein crystallization process, thus increasingly favored by structural biology researchers. Additionally, the rapid freezing technique of samples allows testing in an environment close to natural physiological conditions; sample consumption is minimal, only requiring 0.1 mg, saving researchers valuable protein samples.
Although SPA does not require crystallization, the main issue is that its resolution is difficult to reach the level of X-ray techniques, generally only up to 3 Å. Therefore, some ligands, such as small molecule drugs interacting with target proteins, may not be observable in the electron microscope's density maps; small molecular weight protein particles, due to insufficient contrast, cannot achieve high-resolution images, thus SPA requires that protein samples typically need to be greater than 200 KDa, and small molecular weight samples need to be combined with other proteins to form larger molecular weight samples for testing; testing throughput is low, SPA samples need several days of data collection, generally, each device can only test 1-2 samples per week; SPA has very high hardware requirements, equipment is expensive and maintenance costs are high, and the experience of the experimental personnel is also highly demanded, different experimenters may have significant differences in success rates.
Q2: When should SPA Cryo-EM be chosen?
When there is little experimental material, and it is difficult to crystallize, it is a large biomolecular complex (>150 kDa, up to 2000 Å) that requires observation of the true "native state" structure.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
Cryo-Electron Microscopy Based Service
Protein Structure Identification Service
Protein Structure Identification Service
References
[1] Mackay JP, Landsberg MJ, Whitten AE, Bond CS. Whaddaya Know: A Guide to Uncertainty and Subjectivity in Structural Biology. Trends Biochem Sci. 2017 Feb;42(2):155-167. doi: 10.1016/j.tibs.2016.11.002. Epub 2017 Jan 12. PMID: 28089412.
[2] Rout MP, Sali A. Principles for Integrative Structural Biology Studies. Cell. 2019 May 30;177(6):1384-1403. doi: 10.1016/j.cell.2019.05.016. PMID: 31150619; PMCID: PMC6810593.
[3] Carroni M, Saibil HR. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods. 2016 Feb 15;95:78-85. doi: 10.1016/j.ymeth.2015.11.023. Epub 2015 Nov 27. PMID: 26638773; PMCID: PMC5405050.
[4] Weissenberger G, Henderikx RJM, Peters PJ. Understanding the invisible hands of sample preparation for cryo-EM. Nat Methods. 2021 May;18(5):463-471. doi: 10.1038/s41592-021-01130-6. Epub 2021 May 7. PMID: 33963356.
[5] Zhang L, Tong H, Garewal M, Ren G. Optimized negative-staining electron microscopy for lipoprotein studies. Biochim Biophys Acta. 2013 Jan;1830(1):2150-9. doi: 10.1016/j.bbagen.2012.09.016. Epub 2012 Sep 29. PMID: 23032862; PMCID: PMC3508368.
[6] Danev R, Yanagisawa H, Kikkawa M. Cryo-Electron Microscopy Methodology: Current Aspects and Future Directions. Trends Biochem Sci. 2019 Oct;44(10):837-848. doi: 10.1016/j.tibs.2019.04.008. Epub 2019 May 8. PMID: 31078399.
[7] Fan X, Wang J, Zhang X, Yang Z, Zhang JC, Zhao L, Peng HL, Lei J, Wang HW. Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Nat Commun. 2019 Jun 3;10(1):2386. doi: 10.1038/s41467-019-10368-w. PMID: 31160591; PMCID: PMC6546690.
[8] Nakane T, Kotecha A, Sente A, McMullan G, Masiulis S, Brown PMGE, Grigoras IT, Malinauskaite L, Malinauskas T, Miehling J, Uchański T, Yu L, Karia D, Pechnikova EV, de Jong E, Keizer J, Bischoff M, McCormack J, Tiemeijer P, Hardwick SW, Chirgadze DY, Murshudov G, Aricescu AR, Scheres SHW. Single-particle cryo-EM at atomic resolution. Nature. 2020 Nov;587(7832):152-156. doi: 10.1038/s41586-020-2829-0. Epub 2020 Oct 21. PMID: 33087931; PMCID: PMC7611073.
[9] Su M, Erwin AL, Campbell AM, Pyburn TM, Salay LE, Hanks JL, Lacy DB, Akey DL, Cover TL, Ohi MD. Cryo-EM Analysis Reveals Structural Basis of Helicobacter pylori VacA Toxin Oligomerization. J Mol Biol. 2019 May 3;431(10):1956-1965. doi: 10.1016/j.jmb.2019.03.029. Epub 2019 Apr 5. PMID: 30954575; PMCID: PMC6625667.
[10] Zhang Z, Shigematsu H, Shimizu T, Ohto U. Improving particle quality in cryo-EM analysis using a PEGylation method. Structure. 2021 Oct 7;29(10):1192-1199.e4. doi: 10.1016/j.str.2021.05.004. Epub 2021 May 27. PMID: 34048698.
[11] Olinares PDB, Kang JY, Llewellyn E, Chiu C, Chen J, Malone B, Saecker RM, Campbell EA, Darst SA, Chait BT. Native Mass Spectrometry-Based Screening for Optimal Sample Preparation in Single-Particle Cryo-EM. Structure. 2021 Feb 4;29(2):186-195.e6. doi: 10.1016/j.str.2020.11.001. Epub 2020 Nov 19. PMID: 33217329; PMCID: PMC7867593.
[12] Al-Azzawi A, Ouadou A, Max H, Duan Y, Tanner JJ, Cheng J. DeepCryoPicker: fully automated deep neural network for single protein particle picking in cryo-EM. BMC Bioinformatics. 2020 Nov 9;21(1):509. doi: 10.1186/s12859-020-03809-7. PMID: 33167860; PMCID: PMC7653784.
How to order?

