





Cryo-Electron Microscopy Based Drug Design Service
1. Structure-Based Drug Discovery and Screening
Structural biology has always played a vital role in drug discovery because it provides the most direct and visible atomic-level information on drug targets and can be applied to every step of preclinical drug development, including target identification and design as well as lead compound optimization. Based on atomic resolution information about the active or regulatory sites of target proteins, drug structural design becomes practical.
Structure-based drug design (SBDD), broadly speaking, refers to drug design based on the structures of ligands and receptor proteins; narrowly, it refers to drug design based on receptor structures, that is, designing lead compounds that regulate the receptor or predicting new compound efficacy based on the magnitude of drug effects and structure-activity relationships, based on the 3D structure of the target (broadly the receptor, such as enzymes, receptors, ion channels, antigens, nucleic acids, polysaccharides, etc.) and the principle of molecular recognition (complementarity). Typically, steps in SBDD include structural determination of target proteins, cavity identification, ligand database construction, ligand docking, and lead compound discovery. SBDD is the mainstream mode of drug development today. It is usually combined with a computational-based fast virtual screening of large chemical libraries, which lowers the cost of the initial screenings. It can directly analyze the binding energy between drugs and targets, improve the hit rate of drug discovery, and assist rational drug design.
Many drugs have reached the market through SBDD technology, such as captopril, an angiotensin-converting enzyme (ACE) inhibitor developed by Bristol Myers Squibb, which was the first to use enzyme-inhibitor structural information; saquinavir, which targets the human immunodeficiency virus protease, developed by Roche; zanamivir, which is the neuraminidase inhibitor developed by Biota; and the most famous breakpoint cluster region-proto-oncogene tyrosine-protein kinase inhibitor, imatinib, developed by Novartis for the treatment of leukaemia. However, the shortcomings of SBDD are also obvious, and obtaining high-resolution and complete structural information on protein targets is a prerequisite for SBDD. Most of the target structures in SBDD are provided by X-ray crystallography. However, for membrane proteins and macromolecular complexes, obtaining high-quality crystal structures is particularly difficult.
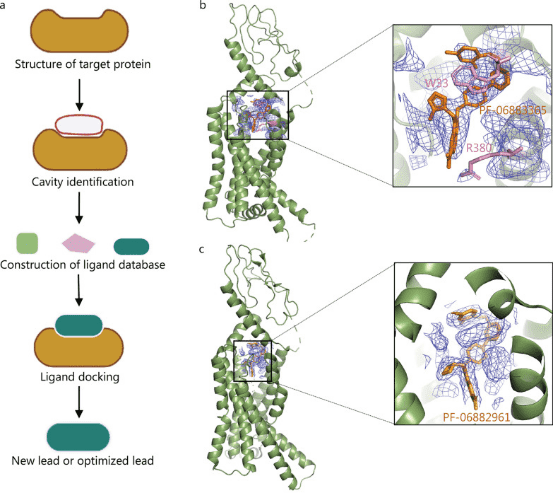
Figure 1. Application of Cryo-EM in SBDD [1]
2. Fragment-Based Drug Discovery
Fragment-Based Drug Design (FBDD) is a new method of drug discovery that combines high-throughput screening with SBDD. The starting materials for screening are smaller molecules, which have a greater degree of freedom and possibility to bind to receptors, which is advantageous. However, because they only bind to partial sites of the receptor, the activity shown is weaker than that of more complex molecules with larger molecular weights, which is an area for improvement. Low-activity fragment molecules can use structural features of binding to the receptor to guide the addition or connection of groups or fragments that help enhance activity, while controlling the size of the molecule to form compounds with potential for drug development.
FBDD has become a mainstream approach in lead compound discovery, encompassing target structure determination, fragment screening, and fragment linking and modification of these fragments. NMR, surface plasmon resonance, and other technologies can be used to screen for small-molecule fragments that have weak interactions with target proteins and then to optimize and connect active fragments based on their structural information to design lead compounds with higher activity. Unlike high-throughput screening to find macromolecules that fit multiple active pockets at the same time, FBDD aims to find small fragments that fit a single active pocket. This approach requires a smaller library of compounds and is notably tolerant of pocket depth variations, accommodating even near-flat protein interaction pockets. FBDD is highly effective but relies heavily on the availability of 3D structural information of target proteins, with purified proteins being essential for this process. Similar to SBDD, the acquisition of 3D structures is often the main factor limiting the development of FBDD technology. In principle, FBDD technology is mainly applicable to proteins with multiple active sites and high molecular mass. Using traditional crystal diffraction techniques to analyze the structure of suitable targets for FBDD is challenging. Therefore, despite over 20 years of development, only six FBDD drugs have been approved for marketing: vemurafenib, venetoclax, erdafitinib, pexidartinib, sotorasib, and asciminib.
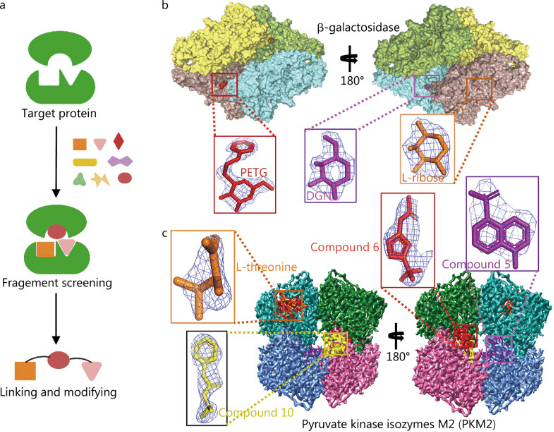
Figure 2. Application of Cryo-EM in FBDD [1]
3. Structural Information Analysis of the Active Ligand and Intermediate Linkers of PROTAC
Proteolysis targeting chimera (PROTAC) is a drug development technology that uses the ubiquitin-proteasome system to degrade target proteins. PROTAC drugs resemble dumbbell structures, consisting of three parts: an E3 ubiquitin ligase ligand, a target protein ligand, and a special linker that connects the two active ligands. When PROTAC drugs enter the patient's body, the target protein ligand and E3 ubiquitin ligase ligand bind to their corresponding proteins, thereby recruiting the E3 ubiquitin ligase near the target protein and causing the target protein to be ubiquitinated, leading to proteasomal degradation of the target protein. PROTAC technology not only accesses some difficult accessible binding sites but also has the advantages of small dosage, high selectivity, and overcoming drug resistance, as it can eliminate overexpressed or mutated targets. The design of PROTAC's active ligands and intermediate linkers highly depends on the structural information of the acting complex. Cryo-EM technology can provide the required structural information.
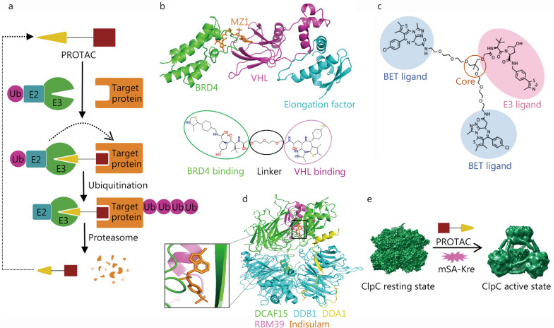
Figure 3. Cryo-EM in PROTAC Development [1]
4. Monoclonal Antibody Epitope Identification
Shortening the screening and structural optimization time for effective antibodies is a long-term goal of antibody development strategies. A recent study reported that combining cryo-EM and next-generation sequencing can rapidly obtain critical epitope information without isolating monoclonal antibodies, significantly shortening antibody development time. After immunizing animals or humans with labeled antigens to obtain antigen and polyclonal antibody complexes, cryo-EM is used to analyze the complex structure to confirm key epitopes and establish an epitope model. Considering the complexity of manually matching density maps with amino acids, studies have also developed an algorithm tool based on density map recognition of antibody sequences, which is matched and scored with a next-generation sequencing database of antigen-binding specific B cells, thus rapidly obtaining epitope information and monoclonal antibody structural models. Recently, cryo-EM has also resolved the structure of B cell receptors, promoting the development of antibody-based therapies. Cryo-EM can guide and promote antibody drug screening, epitope research, and engineering transformation, and is an effective tool for human health research.
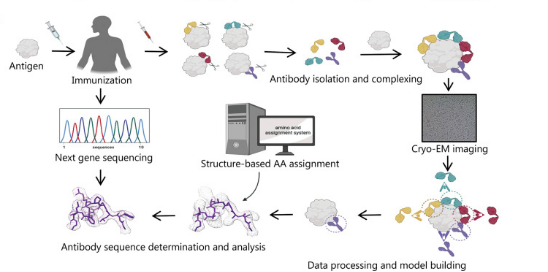
Figure 4. Cryo-EM in Antibody Drug Development [1]
5. Structural Analysis of Enzymes or Enzyme-Substrate Complexes
Recognizing the structural characterization of biomacromolecules also involves defining their dynamic properties, which is one of the important results of characterizing more and more biomacromolecules at high resolution. In fact, early NMR studies of proteins showed that fluctuations occur even within the apparently highly ordered structures, and molecular dynamics simulations indicate that protein structures are undergoing various types of movements at different time scales and amplitudes. Some dynamic properties of macromolecules are crucial for their functional roles, such as pioneering studies on hemoglobin that showed its key ability to transition between different conformational states, thereby more effectively absorbing and releasing oxygen than rigid structures. This dynamic event is increasingly important in many different types of biological processes, from enzyme catalysis to the action of molecular motors, such as those involved in controlling complex biological functions in various life forms, such as actin and myosin.
The presence of extensive dynamic behaviors and multiple conformations poses significant challenges in determining the high-resolution structures of macromolecules in specific functional states. Cross-linking mass spectrometry (XL-MS), small-angle X-ray scattering (SAXS), Förster resonance energy transfer (FRET) between fluorophores, and cryo-EM are the main methods for studying protein dynamics. Cryo-EM is often used to resolve the structures of enzymes or enzyme-substrate complexes, and is particularly useful for larger enzymes or membrane-bound enzymes, capable of solving cryo-EM structures under various conditions.
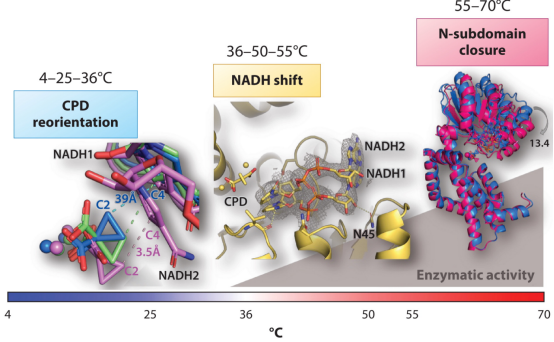
Figure 5. Structural Characterization of the Ternary Complex KARI: 2Mg with Temperature Variations [2]
Analysis Workflow
1. Experimental Procedure Determination Based on Requirements
2. Sample Preparation
3. Data Collection
4. Data Processing
Service Advantages
1. Customizable Experimental Methods Based on Requirements
2. Multi-Gradient Design for Optimal Condition Exploration and Sample Optimization Guidance
3. Automated Equipment Combined with High-Performance Detection Systems Making Experiments Faster, Cheaper, and Simpler
4. Complete Workflow Provided to Achieve 3D Reconstruction
Sample Results
1. Activation Mechanism of PINK1
Protein kinase PINK1 mutations lead to mitochondrial autophagy defects and autosomal recessive early-onset Parkinson's disease. PINK1 possesses several unique functionalities, enabling it to phosphorylate ubiquitin and Parkin's ubiquitin-like domain. Structural analyses of PINK1 from various insect species, with and without ubiquitin, provide snapshots of different structural states, yet fail to elucidate PINK1 activation. Studies employing crystallography and cryo-EM have elucidated the activation mechanism of PINK1. The crystal structure of unphosphorylated Pediculus humanus corporis (Ph; human body louse) PINK1 reveals the N-terminal helix, unveiling the orientation of active yet unphosphorylated PINK1 on mitochondria. Further investigations capture the symmetric PhPINK1 dimer during trans-autophosphorylation and the cryo-EM structure of phosphorylated PhPINK1 undergoing conformational changes to an active ubiquitin kinase state. Structural and phosphorylation studies further delineate the role of PINK1 oxidation regulation. In summary, research delineates the complete activation mechanism of PINK1, elucidating its interaction with the mitochondrial outer membrane and revealing the regulation of PINK1 activity by mitochondrial reactive oxygen species.
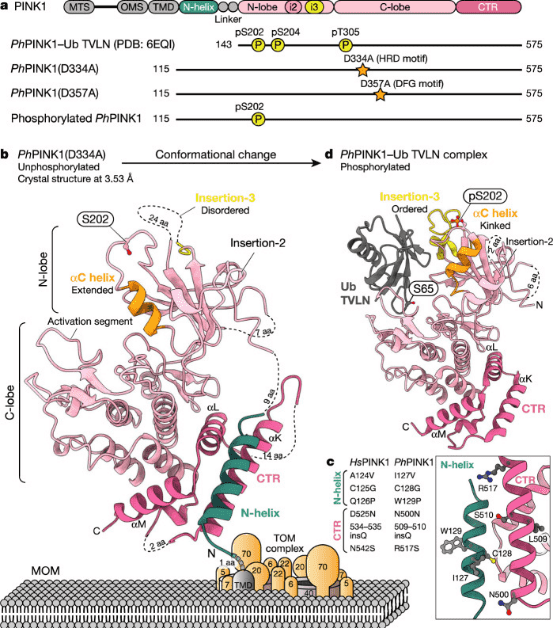
Figure 6. Crystal Structure of the Cytoplasmic Portion of PhPINK1 [3]
2. Cryo-EM Structure of K-Bound hERG Channel Complexed with the Blocker Astemizole
The hERG channel is a voltage-gated potassium channel involved in cardiac repolarization. Inhibition of off-target hERG by drugs has become a critical issue in the pharmaceutical industry. Recently, the 3D structure of the hERG channel was reported using cryo-EM at 3.8-Å resolution. However, due to the scarcity of structural information on the drug- and potassium-bound hERG channel, the mechanism of drug inhibition remains unclear. In this study, the cryo-EM density map of the potassium-bound hERG channel complexed with amiodarone, a well-known hERG blocker associated with an increased risk of potentially fatal arrhythmias, was obtained at 3.5-Å resolution. The structure reveals that astemizole inhibits potassium conduction by directly binding beneath the selectivity filter. Additionally, a possible binding model of astemizole with the hERG channel is proposed, providing insights into hERG's abnormal sensitivity to several drugs.
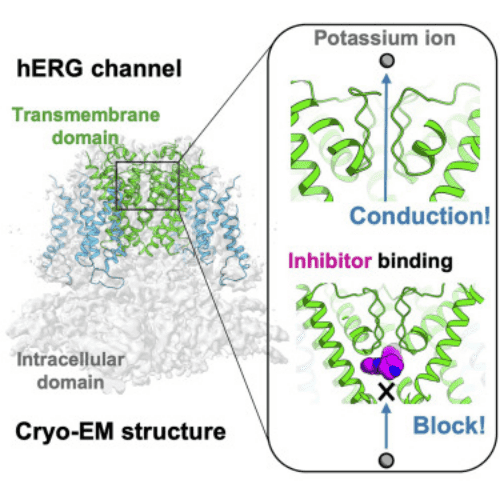
Figure 7. Binding of K-bound hERG Channel with Astemizole Complex [4]
3. Cryo-EM Structure of Gonococcal Multidrug Efflux Pump Elucidates Drug Recognition and Resistance Mechanisms
Neisseria gonorrhoeae has emerged as a highly antimicrobial-resistant Gram-negative pathogen. Multidrug efflux is a primary mechanism employed by N. gonorrhoeae to counteract the effects of multiple classes of antibiotics. It appears that gonococci bearing mosaic-like sequences within the gene mtrD , encoding the most predominant and clinically important transporter of any gonococcal multidrug efflux pump, significantly elevate drug resistance and enhance transport function. Cryo-EM structure of N. gonorrhoeae MtrD, with the mosaic-like sequence, provides insights into the mechanism of drug recognition. This study will ultimately provide information for structure-guided drug design to inhibit these critical multidrug efflux pumps.
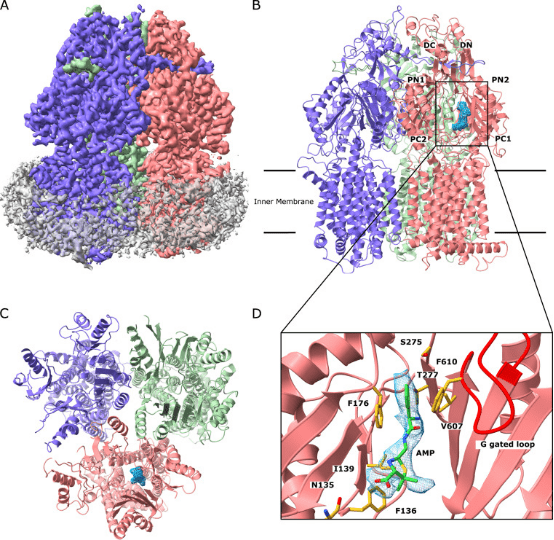
Figure 8. Cryo-EM Structure of MtrD [5]
4. Structural Basis of Sofosbuvir and Amiodarone's Severe Adverse Effects on L-Type Cav Channels
It has been reported that the antiviral drug sofosbuvir and the antiarrhythmic drug amiodarone interact to cause fatal heartbeat slowing. Sofosbuvir and its analog MNI-1 enhance amiodarone's inhibitory effect on cardiomyocyte calcium handling. Although amiodarone is a multi-channel antagonist, its inhibition mechanism on L-type Cav channels remains unclear. A study proposed a systematic cryo-EM structural analysis of Cav1.1 and Cav1.3 alone, treated with amiodarone or sofosbuvir, or sofosbuvir/MNI-1 combined with amiodarone. While amiodarone occupies the dihydropyridine binding site alone, sofosbuvir is not detected in the channel when used alone. In the presence of amiodarone, sofosbuvir/MNI-1 anchors in the central cavity of the pore domain through specific interactions with amiodarone, directly obstructing ion permeation pathways. The study elucidates the molecular basis for the physical, pharmacodynamic interaction of two drugs on the scaffold of Cav channels.
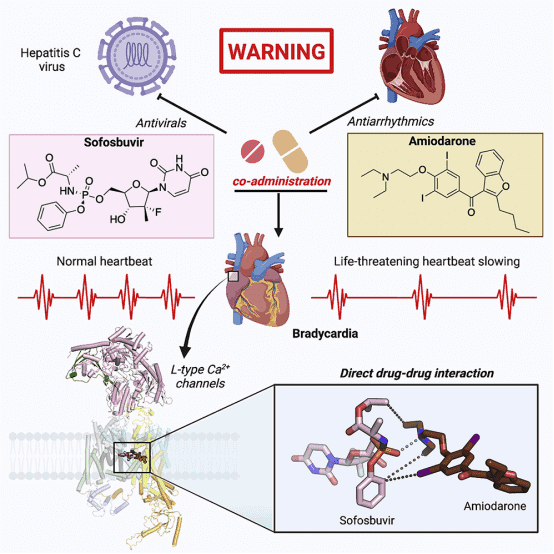
Figure 9. Molecular Basis for the Physical, Pharmacodynamic Interaction of Two Drugs on the Scaffold of Cav Channels [6]
Sample Submission Requirements
1. Comprehensive Experimental Steps
2. Specifications of Relevant Instrumentation
3. Original Experimental Data
4. Data Analysis Report
Applications
1. Cryo-EM in Materials Science and Nanoscience
Cryo-EM laid the foundation for the 2017 Nobel Prize in Chemistry for its profound impact on the field of structural biology by freezing and stabilizing fragile biomolecules for near atomic-resolution imaging in their native states. Beyond the life sciences, the development of cryo-EM for physical sciences could provide previously unattainable length scales for material characterization within systems. Fundamental breakthroughs in discovery and understanding could ensue. Six key areas in materials science that may benefit from the interdisciplinary application of cryo-EM include: (1) batteries, (2) soft polymers, (3) metal-organic frameworks, (4) perovskite solar cells, (5) electrocatalysts, and (6) quantum materials. Addressing long-standing issues in these fields with cryo-EM will firmly establish this powerful tool's broad scope and practicality beyond biology.
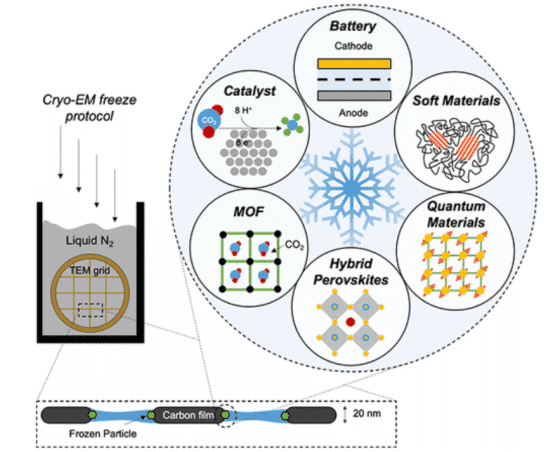
Figure 10. Application of Cryo-EM in Materials Science and Other Fields [7]
FAQ
Q1: What are the advantages of Cryo-EM SPA?
Unlike optical microscopes with a resolution of 0.2 μm, transmission electron microscopes offer a resolution of 0.2 nm, providing a 1000-fold enhancement; (2) Electron tomography enables the study of the internal structure of subcellular components in their native states without requiring protein crystallization; (3) Cryo-EM SPA is suitable for studying large molecules with symmetrical structures as well as irregularly structured large molecular complexes. There is no upper limit on molecular weight; theoretically, molecules >100 kD can achieve sufficient contrast for image correction under ensured imaging conditions.
References
[1] Zhu KF, Yuan C, Du YM, Sun KL, Zhang XK, Vogel H, Jia XD, Gao YZ, Zhang QF, Wang DP, Zhang HW. Applications and prospects of cryo-EM in drug discovery. Mil Med Res. 2023 Mar 6;10(1):10. doi: 10.1186/s40779-023-00446-y. PMID: 36872349; PMCID: PMC9986049.
[2] Tsai MD, Wu WJ, Ho MC. Enzymology and Dynamics by Cryogenic Electron Microscopy. Annu Rev Biophys. 2022 May 9;51:19-38. doi: 10.1146/annurev-biophys-100121-075228. Epub 2021 Dec 21. PMID: 34932913.
[3] Gan ZY, Callegari S, Cobbold SA, Cotton TR, Mlodzianoski MJ, Schubert AF, Geoghegan ND, Rogers KL, Leis A, Dewson G, Glukhova A, Komander D. Activation mechanism of PINK1. Nature. 2022 Feb;602(7896):328-335. doi: 10.1038/s41586-021-04340-2. Epub 2021 Dec 21. Erratum in: Nature. 2022 Mar;603(7903):E33. PMID: 34933320; PMCID: PMC8828467.
[4] Asai T, Adachi N, Moriya T, Oki H, Maru T, Kawasaki M, Suzuki K, Chen S, Ishii R, Yonemori K, Igaki S, Yasuda S, Ogasawara S, Senda T, Murata T. Cryo-EM Structure of K+-Bound hERG Channel Complexed with the Blocker Astemizole. Structure. 2021 Mar 4;29(3):203-212.e4. doi: 10.1016/j.str.2020.12.007. Epub 2021 Jan 14. PMID: 33450182.
[5] Lyu M, Moseng MA, Reimche JL, Holley CL, Dhulipala V, Su CC, Shafer WM, Yu EW. Cryo-EM Structures of a Gonococcal Multidrug Efflux Pump Illuminate a Mechanism of Drug Recognition and Resistance. mBio. 2020 May 26;11(3):e00996-20. doi: 10.1128/mBio.00996-20. PMID: 32457251; PMCID: PMC7251214.
[6] Yao X, Gao S, Wang J, Li Z, Huang J, Wang Y, Wang Z, Chen J, Fan X, Wang W, Jin X, Pan X, Yu Y, Lagrutta A, Yan N. Structural basis for the severe adverse interaction of sofosbuvir and amiodarone on L-type Cav channels. Cell. 2022 Dec 8;185(25):4801-4810.e13. doi: 10.1016/j.cell.2022.10.024. Epub 2022 Nov 22. PMID: 36417914; PMCID: PMC9891081.
[7] Li Y, Huang W, Li Y, Chiu W, Cui Y. Opportunities for Cryogenic Electron Microscopy in Materials Science and Nanoscience. ACS Nano. 2020 Aug 25;14(8):9263-9276. doi: 10.1021/acsnano.0c05020. Epub 2020 Jul 28. PMID: 32806083.
How to order?

