





Comparison of Protein Quantification Methods: iTRAQ, SILAC, AQUA, and Label-Free Quantification Techniques
-
Enables quantification of up to 4 or 8 samples in parallel (depending on the iTRAQ reagent kit)
-
Offers high reproducibility, making it suitable for large-scale proteomics studies
-
Compatible with complex biological matrices, including tissues and body fluids
-
High cost of labeling reagents and complexity of sample preparation
-
Susceptible to ratio compression, which can compromise quantification accuracy
-
Requires high-resolution MS platforms such as Orbitrap or Q-TOF for optimal performance
-
High labeling efficiency, minimizing inter-sample variability
-
Maintains conditions close to physiological environments, eliminating the need for chemical labeling
-
High quantitative accuracy, well-suited for studying dynamic changes in protein expression
-
Applicable only to cell lines that are amenable to in vitro culture
-
Relatively expensive and requires specialized laboratory conditions
-
Limited multiplexing capacity, typically restricted to three labeling states (light, medium, and heavy)
-
Cost-effective and operationally simple, as it eliminates the need for labeling reagents
-
High-throughput capability, ideal for large-scale or exploratory studies
-
Compatible with a wide range of mass spectrometry platforms
-
Requires high instrument stability and reproducibility across runs
-
Offers relatively lower quantitative precision than labeling-based approaches
-
Involves complex data analysis workflows, necessitating advanced computational tools
-
Enables precise absolute quantification
-
Exhibits high specificity, ideal for validation studies
-
Well-suited for detecting low-abundance proteins or biomarkers
-
Requires the synthesis of isotope-labeled peptides, leading to high cost
-
The number of quantifiable target proteins is limited
-
Not appropriate for high-throughput screening
In contemporary life science research, protein quantification serves as a crucial tool for elucidating biological mechanisms, identifying disease biomarkers, and investigating drug mechanisms of action. Compared to conventional protein expression analysis techniques such as Western blotting and ELISA, mass spectrometry (MS)-based quantitative proteomics offers distinct advantages, including high throughput, superior sensitivity, and strong multiplexing capabilities. As a result, it has become a core methodology in proteomics research.
Currently, mainstream strategies for protein quantification fall into four primary categories: iTRAQ, SILAC, Label-Free, and AQUA. Each technique is characterized by distinct principles and application suitability. This paper systematically compares the underlying mechanisms, strengths, and limitations of these four methods to assist researchers in selecting the most appropriate approach for their experimental needs.
iTRAQ: A Label-Based High-Throughput Quantification Strategy
📌Technical Principle
iTRAQ (Isobaric Tags for Relative and Absolute Quantitation) is an isotope labeling method that quantifies peptides across multiple samples by chemically tagging their amine groups with isobaric reagents. These tags are indistinguishable in MS1 spectra, appearing as identical precursor ions, but are differentiated in MS2 by their release of unique reporter ions, allowing for simultaneous quantification of multiple samples.
📌Advantages
📌Limitations
📌Application Scenarios
Well-suited for high-throughput comparative studies, including disease proteomics and pharmacodynamic profiling, where simultaneous analysis of multiple samples is essential.
SILAC: Isotope Labeling Technique at the Cellular Level
📌Technical Principle
Stable Isotope Labeling by Amino acids in Cell culture (SILAC) involves the incorporation of “light” or “heavy” isotopically labeled amino acids (e.g., ^13C- or ^15N-labeled lysine or arginine) into proteins during cell growth. These labeled amino acids are metabolically incorporated during protein synthesis, enabling proteins to be labeled at the expression stage. The samples can then be directly subjected to LC-MS/MS analysis to quantify differential protein expression under various treatment conditions.
📌Advantages
📌Limitations
📌Application Scenarios
SILAC is particularly suitable for mechanistic investigations such as elucidating intracellular signaling pathways and drug action mechanisms.
Label-Free: Simple Strategy without Labeling
📌Technical Principle
Label-Free Quantification (LFQ) performs relative quantification based on peptide ion intensity (e.g., peak area) or spectral counting, without requiring any isotopic or chemical labeling. This method allows the analysis of an unrestricted number of samples and is highly scalable for large datasets.
📌Advantages
📌Limitations
📌Application Scenarios
Label-Free quantification is especially well-suited for early-stage exploratory research and high-throughput screening of clinical samples involving large cohorts.
AQUA: The Gold Standard for Absolute Quantification
📌Technical Principle
AQUA (Absolute QUAntification) employs synthetic peptides labeled with stable isotopes at known concentrations as internal standards. These peptides are spiked into the sample and analyzed via LC-MS/MS. The absolute quantity of the target protein is then determined using a calibration curve.
📌Advantages
📌Limitations
📌Application Scenarios
AQUA is widely employed in biomarker validation and the development of clinical diagnostic methods.
Summary of Technical Comparison
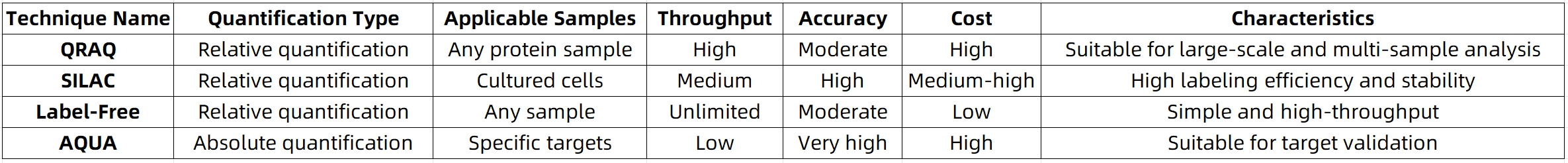
MtoZ Biolabs is dedicated to delivering high-quality quantitative proteomics services. Leveraging cutting-edge mass spectrometry platforms and a team of experienced analysts, we design optimized experimental workflows tailored to client needs and provide comprehensive data mining and interpretation of analytical results.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

