





Advanced Mass Spectrometry Enables Single-Cell Insights: proteoCHIP EVO 96 Accelerates Single-Cell Proteomics
Single-cell proteomics (SCP) represents a forefront area of biological research, dedicated to profiling protein expression patterns and their dynamics at the single-cell level. Traditional proteomics methods predominantly focus on population or tissue-scale analyses, limiting their capacity to resolve cellular heterogeneity. However, such heterogeneity is critical for understanding tissue functionality, biological mechanisms, and disease progression, underscoring the need for precise single-cell detection technologies. In recent years, mass spectrometry-based single-cell proteomics has achieved notable advancements, with improvements in detection sensitivity and throughput. Despite these developments, reproducibly quantifying thousands of proteins within individual cells remains a significant challenge. To address this limitation, Steven A. Carr and Claudia Ctortecka from the Broad Institute of MIT and Harvard introduced a specialized sample preparation platform, proteoCHIP EVO 96, in combination with the Evosep One liquid chromatography system. This integration enables an efficient, high-throughput, and low-loss single-cell proteomics workflow. The application of proteoCHIP EVO 96 technology not only propels advancements in single-cell proteomics but also offers innovative tools for addressing complex biological questions. As a provider of high-quality biological mass spectrometry services, MtoZ Biolabs is committed to delivering advanced, comprehensive single-cell proteomics analysis solutions to the scientific community.
The novel workflow based on proteoCHIP EVO 96 significantly enhances protein detection depth and throughput by directly transferring single-cell samples to the Evosep system for online desalting and separation. This approach demonstrates its capacity to analyze complex biological processes at the single-cell level. Additionally, the technology supports fully automated operation, streamlining single-cell sample preparation and injection workflows. In July 2024, the team from the Broad Institute of MIT and Harvard published their key findings and technical advancements in Nature Communications:
1. Minimization of Peptide Adsorption Loss by proteoCHIP EVO 96
ProteoCHIP EVO 96 is a micromachined low-adsorptive PTFE chip featuring tapered nanopores in a 96-well plate layout. Each nanopore is preloaded with 3 μL of hexadecane. Protein extraction and enzymatic digestion occur directly within these nanopores, minimizing peptide loss associated with pipetting and sample transfer. By adjusting the temperature of proteoCHIP EVO 96, liquid droplets separate from hexadecane, aligning with Evotip cartridges for downstream Evosep One chromatography. Using both manual and automated sample injection with five single-cell samples, the study evaluated peptide recovery efficiency (Figure 1a). Manual injection yielded a median of 582 proteins per analysis, while automated Evotips injection achieved 812 proteins per single cell. Although manual injection did not miss peptides with specific hydrophobic indices (GRAVY), automated injection detected lower-abundance peptides (Figure 1c). The peptide sequence overlap between the two strategies reached 99%, with only 37 unique peptides observed in automated samples, demonstrating the efficiency of proteoCHIP EVO 96's specialized design.
Optimizing the default ddaPASEF acquisition strategy (Figure 1b, c), the integration of direct sample loading with the 40SPD Evosep One method nearly doubled protein identification and increased sample throughput by 30%.
Further comparisons revealed a 12% improvement in data completeness with direct Evosep injection compared to manual nanoElute methods (Figure 1e). Moreover, the dynamic range of peptide quantification remained consistent between both methods, spanning approximately five orders of magnitude (Figure 1f). diaPASEF acquisition demonstrated higher precursor abundance in direct Evosep SCP samples than in manual nanoElute samples (Figure 1c vs. f). These findings suggest that combining a dedicated diaPASEF acquisition strategy with the peak capacity of 40SPD enhances precursor sampling efficiency across the entire dynamic range (Figure 1f).
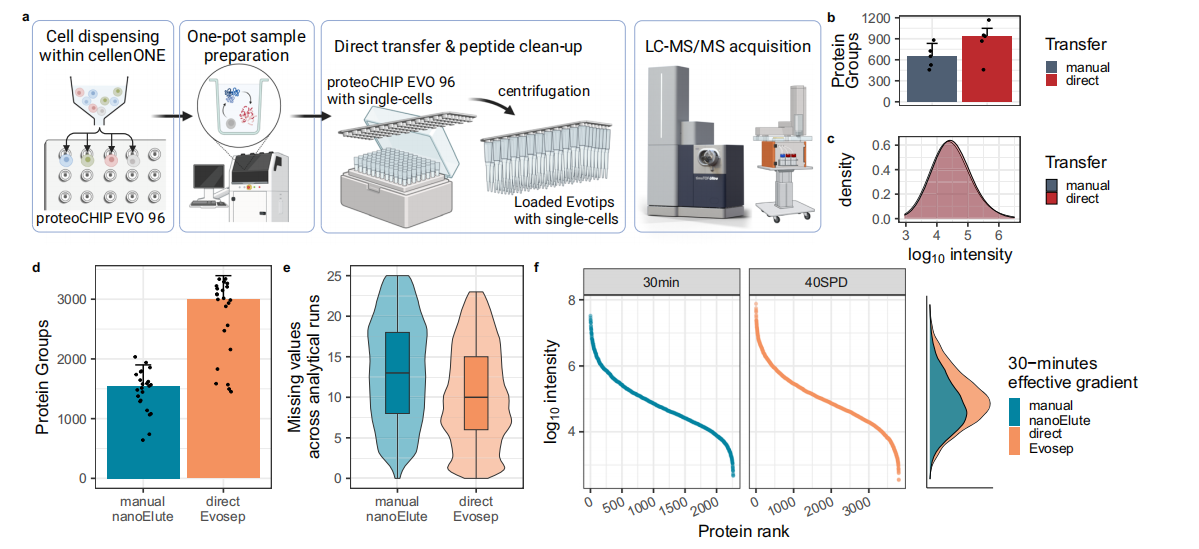
Figure 1. Comparison of Direct vs. Manual Sample Injection
2. Up to 3,500 Proteins Can Still Be Identified Under High-Throughput Conditions
Standard single-cell proteomics (SCP) projects often require the analysis of hundreds to thousands of single cells to capture tissue or population-level heterogeneity and to uncover underlying biological mechanisms. However, reducing chromatographic separation time introduces challenges related to acquisition speed and ion utilization efficiency in timsTOF-based SCP workflows. In this study, peptide separation was performed at a flow rate of 100 nL/min using both 40SPD and 80SPD modes on a 5 cm Aurora Rapid column. The 80SPD method doubled the throughput compared to 40SPD, and the results revealed that nearly all proteins identified with the 80SPD approach were also detected using the 40SPD method. This reproducibility highlights the robustness of the SCP workflow and acquisition strategy developed by the research team (Figure 2b).
Subsequently, the total abundance of quantified proteins was analyzed using both methods. The increased throughput achieved with 80SPD resulted in a compressed dynamic range for single-cell analysis, reducing it by approximately one order of magnitude (Figure 2c). To evaluate the impact of this reduction on protein identification and quantification, E3 ubiquitin ligases were chosen as target proteins. With the 40SPD method, over 50 E3 ubiquitin ligases were identified in single cells, whereas 13 were detected using the 80SPD approach (Figure 2c). Although the signal intensity of the ligases identified via the 80SPD method decreased by nearly one order of magnitude, the relative ranking of protein abundances remained consistent between the two approaches (Figure 2c). Despite the reduced number and dynamic range of proteins identified with the 80SPD method, the identified proteins largely overlapped with those detected by 40SPD. This suggests that the proteins missed in the 80SPD dataset were likely of low abundance. Indeed, proteins with MS1 abundance below 1e3 in the 40SPD dataset generally fell below the detection limit in the 80SPD analysis (Figure 2d). Additionally, more frequent precursor co-elution in the shorter chromatographic gradient increased the complexity of MS/MS scans, potentially suppressing the signals of low-abundance precursors (Figure 2e). These findings indicate that while increased throughput comes at the cost of reduced analysis depth, a twofold improvement in single-cell proteomics throughput is technically achievable.
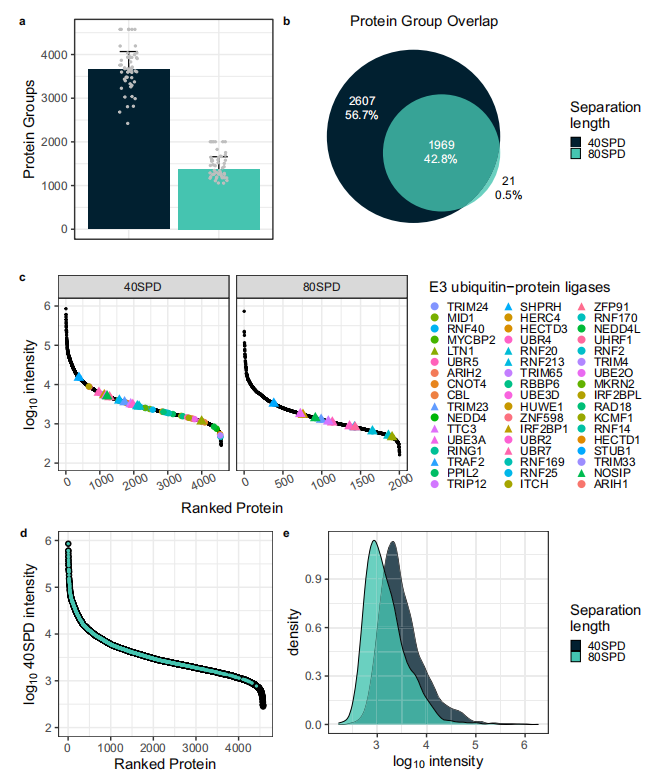
Figure 2. Impact of Increased Throughput on Protein Identification Results
3. Validation of LPS-Induced Proteomic Changes at the Single-Cell Level
Lipopolysaccharide (LPS) is known to stimulate inflammation, induce cytokine production, and activate metabolic responses across various cell types. This study investigated whether the proteoCHIP EVO 96-based single-cell proteomics (SCP) workflow could effectively analyze the effects of LPS stimulation at the single-cell level. To this end, THP-1 human monocytic leukemia cells (13.7 µm) were treated with 200 ng/mL LPS (n = 77) or DMSO as a control (n = 84) for 12 hours. Following treatment, cells were processed using the proteoCHIP EVO 96 workflow, transferred onto Evotips, and analyzed on a timsTOF Ultra instrument using the 40SPD method (Figure 3a). A total of 1,537 proteins were identified, with a median of 1,149 proteins detected per single cell (Figure 3b). Compared to HEK-293T cells (26.7 µm) shown in Figure 2a, the number of proteins identified per cell was lower, likely due to the smaller cell volume of THP-1 cells (Figure 2a, Figure 3b). Differential protein analysis revealed significant changes in LPS-treated cells compared to DMSO controls. Specifically, 214 proteins were significantly upregulated, while 15 proteins were significantly downregulated (Figure 3d). Further Gene Set Enrichment Analysis (GSEA) indicated enrichment in pathways associated with interferon and interleukin signaling, with a notable emphasis on the interleukin-12 family (Figure 3e). These findings are consistent with the established biological effects of LPS.
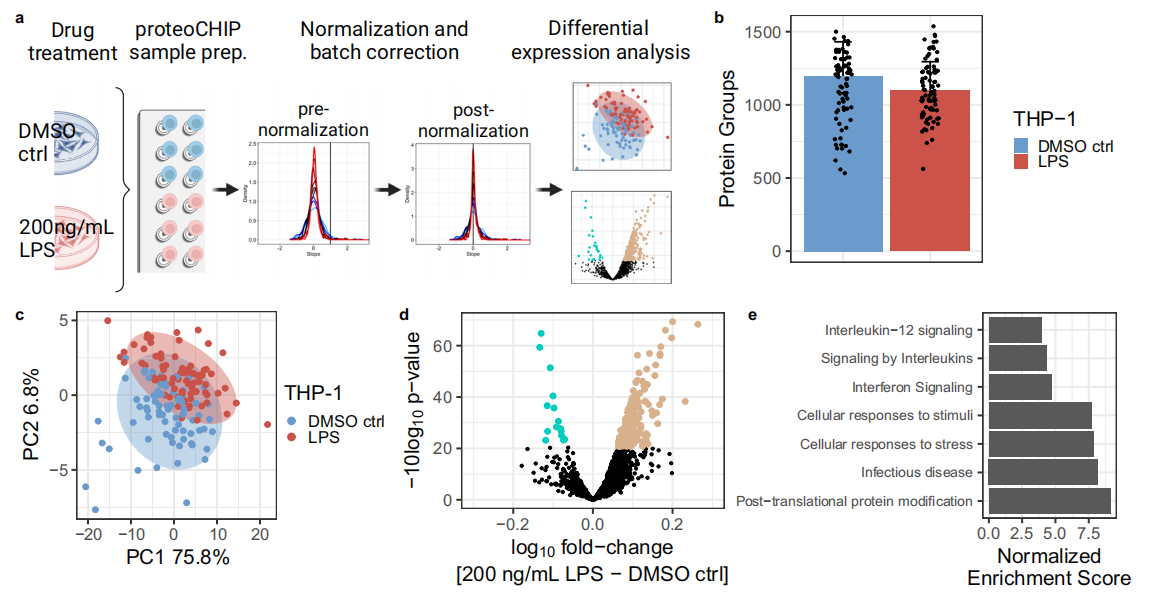
Figure 3. Results of Single-Cell Proteomics Analysis Using the 40SPD Workflow
The study employed specially designed chips, including cellenONE and proteoCHIP EVO 96, for efficient sample processing. By directly interfacing with the Evosep One chromatography system, the workflow achieved online desalting and reproducible peptide separation. Coupled with the Bruker timsTOF system, this automated setup eliminated manual sample handling and ensured consistent performance. Using the latest timsTOF Ultra platform, up to 4,000 proteins were identified, with an average detection of 3,500 proteins per HEK-293T cell. Furthermore, the study analyzed hundreds of LPS-stimulated THP-1 cells using a reproducible single-cell proteomics workflow, identifying key regulatory proteins associated with interleukin and interferon signaling pathways. These findings demonstrate that proteoCHIP EVO 96-based sample preparation, when integrated with timsTOF Ultra, provides sufficient proteome depth to study intricate biological phenomena beyond simple cell-type classification.
ProteoCHIP EVO 96 technology, through its innovative design and seamless integration with the Evosep One chromatography system, improves sample preparation efficiency, detection depth, and analysis throughput while minimizing manual errors. This advancement provides new avenues for single-cell proteomics research. The technology has potential applications across various fields, including investigating cellular functional heterogeneity, exploring inflammation response mechanisms, identifying drug targets in fundamental research, and developing personalized diagnostics and therapies in clinical applications. It offers strong support for uncovering biological complexities and addressing challenging diseases, contributing significantly to advancements in the field of single-cell proteomics.
MtoZ Biolabs provides accurate and efficient single-cell proteomics analysis services by leveraging cutting-edge technologies, including the Thermo Orbitrap Astral high-resolution mass spectrometer, the Bruker timsTOF HT system, and the fully automated single-cell sorting platform cellenONE. Whether conducting high-throughput cellular profiling or performing in-depth analysis of complex biological samples, MtoZ Biolabs ensures reliable data output and tailored solutions, empowering researchers to achieve their scientific goals.
MtoZ Biolabs, an integrated chromatography and mass spectrometry (MS) services provider.
Related Services
How to order?

