





Protein Structures and Interactions Analysis Service | LiP-MS
Limited proteolysis mass spectrometry (LiP-MS) has some of the same mechanisms as drug affinity responsive target stability (DARTS). LiP-MS is a newly developed proteomics approach that combines finite protein breakdown with targeted proteomics workflows. LiP-MS analysis of biological samples under native conditions and limited proteolysis requires protein digestion with proteinase K (proK) and trypsin. Then, the sequentially generated peptides are analyzed by LC-MS. Finally, label-free quantitative proteomic analysis is used to compare the protective effect of the peptide in the control group and drug-treated samples to determine the target protein.
Unlike DARTS, LiP-MS can recognize subtle peptide fingerprints with a resolution of only 12 amino acids to detect small molecule (SM) binding sites, which is useful for identifying missing protein structural information in thermal shift assay. LiP-MS also has the following advantages: (1) it can identify and detect protein-SM interactions, including allosterism, enzyme-substrate, enzyme-drug, and drug-target interactions. (2) In situ structural changes can be detected by it. (3) Peptide fingerprinting can be assessed at the proteome level through it. (4) Structural changes in different proteins can be detected by it under various conditions. However, there are some limitations to this method. First, the binding site can only be determined when the peptide is detected by MS, which requires high sequence coverage of the target protein. In addition, uncertainty in conformational changes may hinder the identification of binding sites.
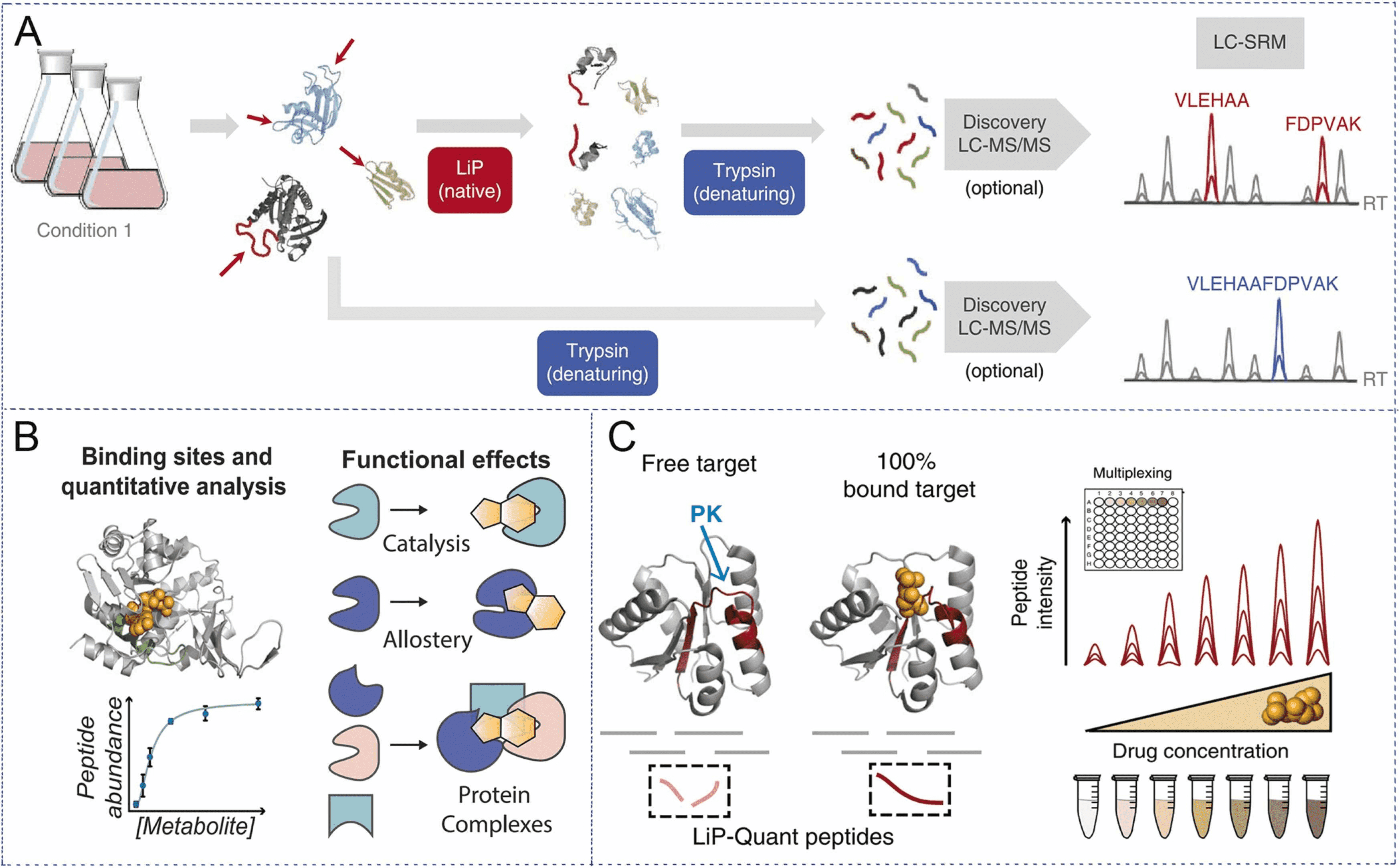
Figure 1. Experimental Principles of LiP-MS [1]
LiP-MS, a technique that performs limited proteolysis with low concentrations of non-selective proK, preferentially cleaves the exposed flexible portion of the protein (looped or unfolded), denaturing and trypsinization, and analyzing peptide mixtures by LC-MS, can identify drug targets in complex biological environments without the need for chemical modification of ligands.
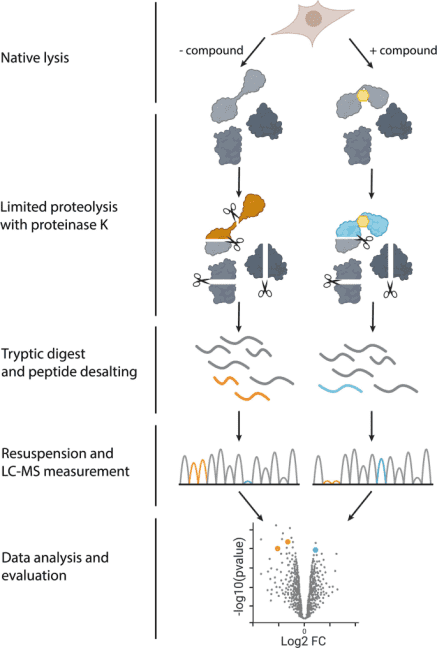
Figure 2. Steps of LiP-MS Experiment [2]
In 2023, Nature Protocol reported a specific experimental method for measuring structural changes at the proteome scale using LiP-MS. LiP-MS consists of five main steps: (1) sample preparation, in which each sample is divided into two aliquots (LiP and trypsin control (TrP) samples); (2) limited proteolysis of LiP samples under native conditions; (3) complete proteolytic digestion of LiP and TrP samples under denaturing conditions; (4) data collection; (5) data analysis and visualization.
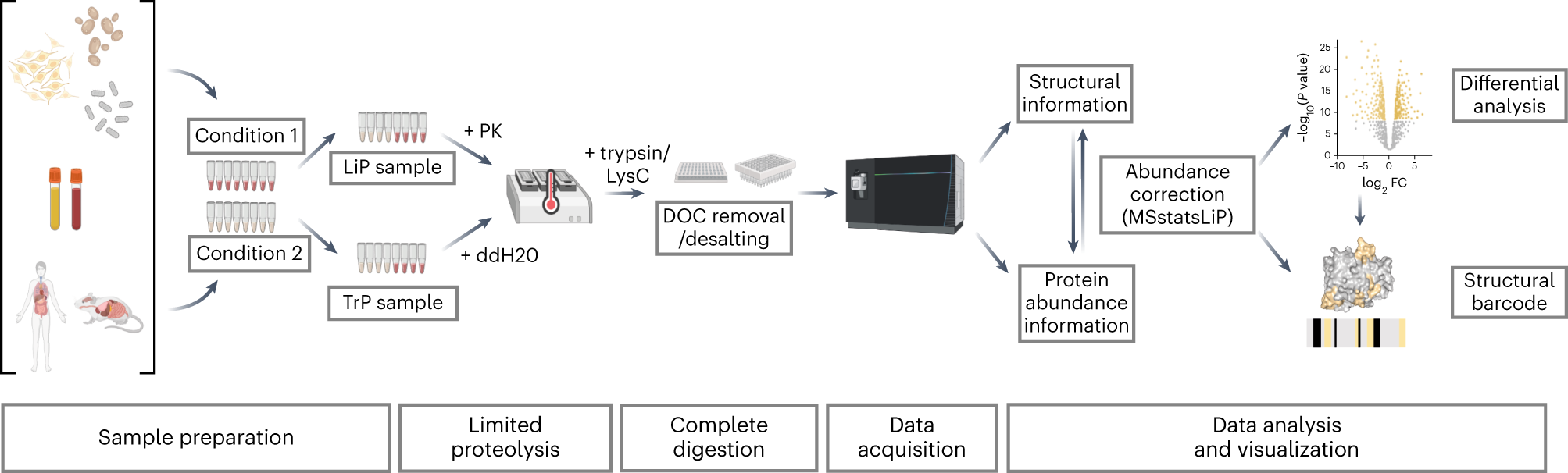
Figure 3. Synthesis of ABPs and AfBPs [3]
1. Sample Preparation
LiP-MS is suitable for a wide range of samples, from purified proteins or complex of whole cell or tissue lysates and body fluids. Therefore, the extraction method is determined by the type of sample, and the method with the highest protein yield should be selected while remaining undenaturing, preferably under physiological conditions, which are most likely to preserve the native protein structure. In cell lysis, endogenous proteases can cause unwanted proteolysis. Therefore, it is recommended to perform the extraction step at low temperature (4℃) with the addition of protease inhibitors to the extraction buffer.
In the LiP-MS experiment, each sample is equally divided into control and experimental groups, each sample is divided into two equal portions, and processed in two parallel experiments. The first aliquot undergoes limited proteolysis with the non-specific proK under native conditions to produce a structure-specific proteolytic pattern, followed by complete digestion with the endoprotease trypsin and Lysyl endopeptidase (LysC) under denaturing conditions (LiP sample). Another aliquot is directly denatured and completely digested to assess protein abundance changes (TrP sample).
2. Limited Proteolysis
Controlled partial digestion of proteins under native conditions is a key step in this method. Depending on the structural state of the protein, the protein's sensitivity to proK limited digestion varies. Therefore, flexible and accessible regions of the protein (such as loops or unstructured domains) will be preferentially targeted by proK, while folding or aggregation reduces PK's accessibility, and the degree of proK cleavage can represent structural flexibility and accessibility. Thus, molecular events that alter protein structure can change sensitivity to proK, resulting in different proteolytic patterns for each protein conformation.
To ensure high reproducibility of the cleavage pattern, the impact of multiple variables on this key proteolytic step must be considered in the experimental design. Particularly, the enzyme/substrate (E:S) ratio and the incubation time of the limited proteolysis step should be strictly controlled. Heating (99℃) followed by denaturation using sodium deoxycholate (DOC) effectively inactivates proK. Complete inactivation of proK is key to the success of the experiment, as residual activity will lead to prolonged digestion time and hinder the generation of structure-specific digestion patterns.
3. Complete Enzymatic Digestion under Denaturing Conditions
In this step, all samples are digested with trypsin and LysC under denaturing conditions to generate peptides suitable for bottom-up MS. First, disulfide bonds are reduced and then alkylated to ensure complete accessibility during digestion. The samples are then digested with protease LysC and trypsin. For LiP samples, prior digestion with proK generates cleavage sites with non-trypsin termini. Peptides containing only one trypsin terminus are called half-trypsin (HT) peptides. For control samples digested only with LysC and trypsin, peptides will have two trypsin termini; these peptides are called full-trypsin peptides. The digestion reaction is stopped by adding formic acid, which causes DOC precipitation. The precipitated DOC is removed, and then the trypsin-digested peptides are desalted using reversed-phase C18 material.
4. Data Collection
DIA is generally used for bottom-up MS detection in LiP-MS, but data-dependent acquisition (DDA) methods and targeted reaction monitoring are also available. After deconvoluting the fragment ion spectra, a protein database can be used for data retrieval in classical proteomics experiments. However, as LiP-MS data includes HT peptides, a study-specific database should typically be generated. This can be achieved by using the DDA method on the same set of samples or by directly searching DIA data. The former can ensure a broader spectrum library, improving the accuracy of identification and quantification, but if all replicate samples of a sample are to be measured, this requires a longer collection time as a cost; this issue can be solved by pooling the various replicate samples in the library generation step.
5. Data Analysis
The main goal of LiP-MS data analysis is to identify proteins that undergo structural changes under target conditions; by comparing samples, it is possible to find out which protein regions are involved in the change. Before performing structural analysis, a variety of quality control measures are used to evaluate the LiP-MS dataset. In short, evaluate the peptide intensity distribution of each run, the coefficient of variation between biological replicates, the proportion of HT peptides in each replicate, and the number of identified proteins and peptides.
LiP-MS data analysis includes differential analysis of peptide intensities between different conditions in LiP samples. Both full-trypsin peptides and HT peptides can be used. Differential analysis is performed using a linear mixed model, which integrates all quantitative information under all conditions, thereby increasing sensitivity. Any contribution from changes in protein abundance is corrected using TrP samples. Proteins that are structurally significantly different between different conditions are identified in the differential analysis of LiP samples. LiP peptides that vary under different conditions are identified based on statistical significance and fold change in intensity. Similarly, in the differential analysis of full-trypsin peptides in TrP samples, proteins with significantly different abundance under the two conditions are identified. The threshold of statistical significance may vary with the experiment.
For data analysis, MSstatsLiP is an R package designed specifically for LiP-MS data analysis, quality assessment, and visualization. MSstatsLiP provides the following functions: data quality control, normalization to minimize systematic variation in MS runs, summarization of spectral features, quantification of LiP peptides and their corresponding proteins, and detection of protein structural changes and/or abundance changes under different experimental conditions. MSstatsLiP offers the possibility of visualizing LiP-MS data as structural barcodes, representing proteolytic fingerprint maps along the protein sequence, and providing a concise visual summary of protein regions, showing conformational changes between conditions. Finally, MSstatsLiP offers a more sophisticated statistical method, based on methods for post-translationally modified data, to correct for abundance changes in LiP-MS data.
Analysis Workflow
1. Determine the Experimental Process According to the Experimental Requirements
2. Pre-Experiment, Optimizing Experimental Conditions
3. Sample Preparation
4. High-Resolution MS Acquisition Data
5. Data Retrieval and Analysis
Service Advantages
1. Identification of Target Proteins from Multiple Types of Sample Sources, etc.
2. Provide Guidance on Optimizing Experimental Conditions
3. High-Confidence, High-Precision MS Detection
4. Comprehensive Bioinformatics Analysis
Sample Results
1. Identification of α-Amanitin Effector Proteins in Hepatocytes by Limited Proteolysis Coupling Mass Spectrometry
The misuse of poisonous mushrooms containing amatoxins can lead to acute liver failure (ALF) in patients and is a cause of a large number of deaths. Although the mechanism of toxicity of α-amanitin (α-AMA) and its interaction with RNA polymerase II (RNAP II) have been studied, the α-AMA effector protein that can interact with α-AMA in hepatocytes has not been systematically studied. Limited proteolysis coupling mass spectrometry (LiP-MS) is an advanced technique based on global comparative proteomics that enables rapid identification of protein-ligand interactions. In this study, the α-AMA effector protein found in human hepatocytes was identified using LiP-MS coupled with tandem mass tag (TMT) technology to detect conformotypic peptides. Based on α-AMA concentration-dependent LiP-MS and LiP-MS in human and mouse hepatocytes, proteins classified as protein processing in the KEGG pathway and ribosomes can be assessed by affinity. The possibility of interaction between α-AMA and proteins containing conformational peptides was evaluated by molecular docking studies. The results of this study suggest that α-AMA induces hepatotoxicity through interaction with various proteins involved in protein synthesis, as well as RNAP II.
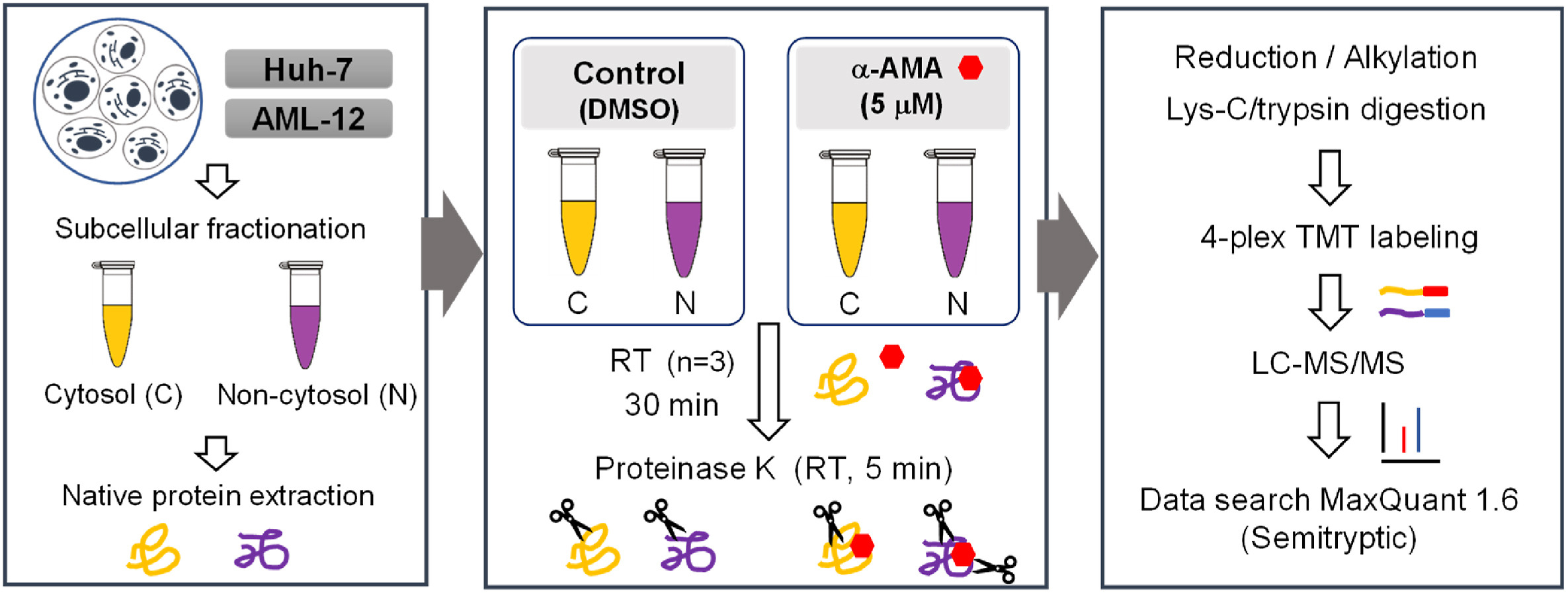
Figure 4. Experimental Ideas for the Application of LiP-MS [4]
2. Identification of Phosphatidylinositol 4,5-Bisphosphate Effectors in the Human Nuclear Proteome by Finite Proteolysis-Coupled Mass Spectrometry
Specific nuclear sub-compartments that are regions of fundamental processes such as gene expression or DNA repair, contain phosphoinositides (PIPs). Thus, PIPs may represent signals for specific proteins to localize to different nuclear functional domains. Limited proteolysis followed by label-free quantitative mass spectrometry was performed and the most abundant nuclear protein effector of PIP, phosphatidylinositol 4,5-bisphosphate (PIP2), was identified. A total of 515 proteins with PIP2 binding capacity were identified, of which 191 'exposed' proteins represented direct PIP2 interactors and 324 'hidden' proteins, where PIP2 binding was increased after trypsin treatment. Gene ontology analysis showed that the 'exposed' protein was involved in gene expression as a regulator of Pol II, mRNA splicing, and cell cycle. They are mainly localized to non-membrane-bound organelles – nuclear speckles and nucleolus – and are attached to the actin nucleoskeleton. 'hidden' proteins have been implicated in gene expression, RNA splicing and transport, cell cycle regulation, and response to heat or viral infection. These proteins localize to the nuclear envelope, nuclear pore complex, or chromatin. Bioinformatics analysis of peptides bound in both groups showed that the PIP2 binding motifs were in general hydrophilic. The data provided insight into the molecular mechanism of nuclear PIP2 protein interactions and advance methods applicable to further studies of PIPs or other protein ligands.
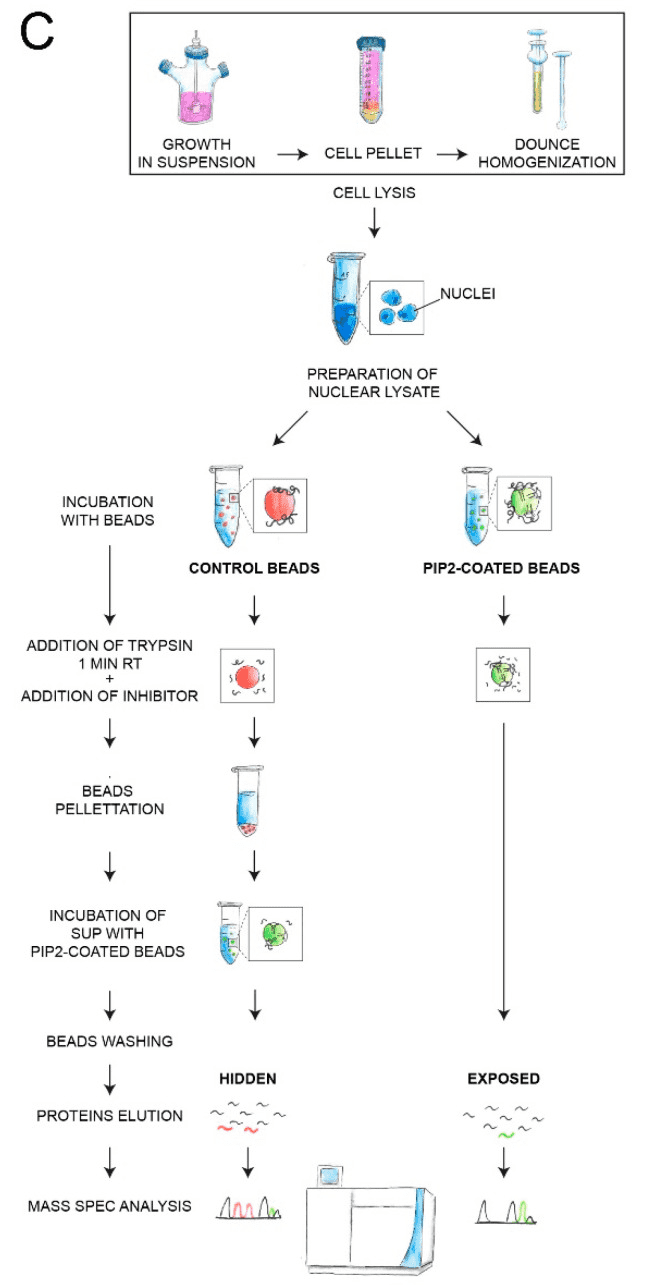
Figure 5. PIPsLiP-qMS Experimental Workflow Protocol [5]
3. The Combination of Phosphate Secretion Profiling Analysis and Kinase Substrate Interaction Screening Defined the Extracellular Signal Transduction Driven by Active c-Src
c-Src tyrosine kinase is a well-known intracellular key signaling molecule and a potential target for cancer therapy. Secreted c-Src is a recent observation, but how it promotes extracellular phosphorylation remains elusive. Using a series of domain-deletion mutants, it has shown that the N-proximal region of c-Src is critical for its secretion. A tissue inhibitor of metalloproteinase 2 (TIMP2) is an extracellular substrate of c-Src. LiP-MS and mutagenesis studies have confirmed that the Src homology 3 (SH3) domain of c-Src and the motif of P31VHP type 34TIMP2 are critical to their interaction. Comparative phosphoproteomic analysis identified the enrichment of the PxxP motif in the phosphorus-containing secretome in c-Src-expressing cells with procarcinogenic effects. Inhibition of extracellular c-Src with a custom SH3-targeting antibody disrupts kinase-substrate complexes and inhibits cancer cell proliferation. These findings point to the intricate role of C-SRC in the production of phosphating secretomes, which may affect cell-to-cell communication, particularly in cancers where C-SRC is overexpressed.
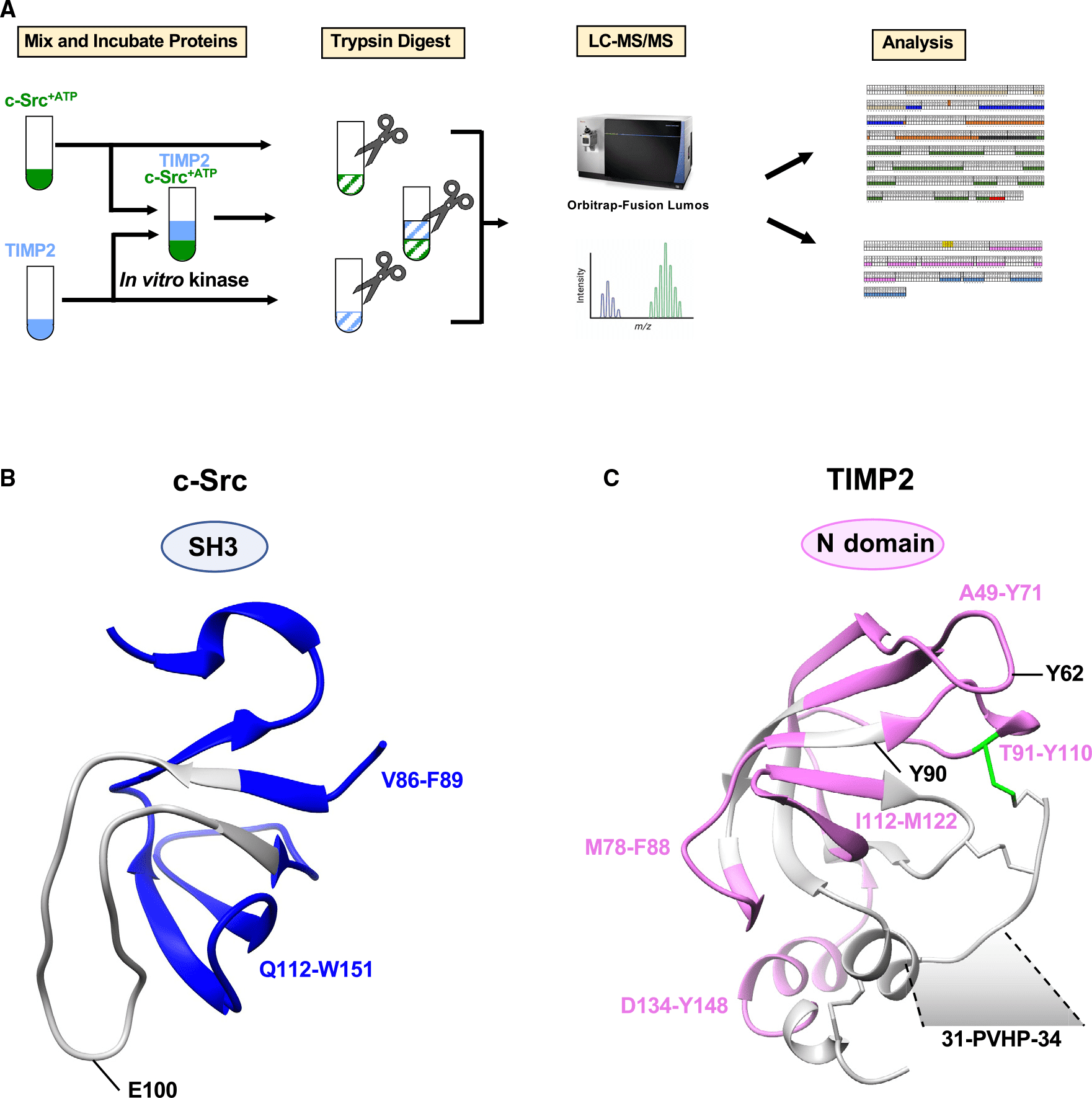
Figure 6. Schematic Diagram of LiP-MS Sample Processing [6]
Sample Submission Requirements
1. Samples such as drugs/proteins should not be contaminated with impurities as much as possible.
Services at MtoZ Biolabs
1. The Complete Experimental Procedure
2. Relevant Instrument Parameters
3. Raw MS Data
4. Data Analysis Reports
Applications
1. Identifying Drug Targets and Binding Sites
Chemical proteomics is a key technique for characterizing the action of a drug, as it directly identifies the protein targets of bioactive compounds and contributes to the development of optimized small molecule compounds. Current methods are unable to identify protein targets for compounds and detect interacting surfaces between ligands and protein targets without prior labeling or modification. To address this limitation, research has developed LiP-Quant, a drug-target deconvolution pipeline based on LiP-MS, applicable across species, including in human cells. Machine learning is used to identify features indicative of drug binding and integrate them into a single score to identify protein targets for small molecules and approximate their binding sites. The study demonstrated drug target identification across compound classes, including drugs targeting kinases, phosphatases, and membrane proteins. LiP-Quant estimates half of the maximum effective concentration of the compound binding site in the whole cell lysate, allowing for the accurate differentiation of drug binding to cognate proteins and the identification of previously unknown targets for biocide research compounds.
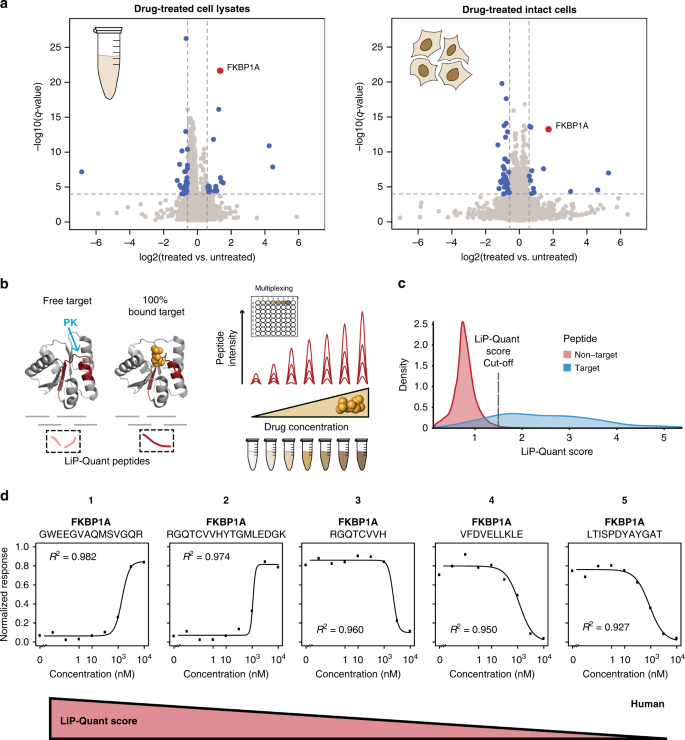
Figure 7. LiP-Quant Experimental Process [7]
FAQ
Q1: What factors affect LiP-MS data acquisition?
LiP-MS is a unique method that can both perform proteome-wide target deconvolution and identify structural changes in the target protein upon compound binding. However, several factors influencing LiP-MS targeted deconvolution should also be considered: (i) adequate sequence coverage is required to detect peptides near compound binding sites, thus LiP-MS tends to favor proteome-rich proteins. (ii) Not all compounds may affect the pharmacokinetic accessibility of the target. (iii) In addition to influencing the structure of the binding site, small molecules may also cause allosteric changes outside of that site. Since both of these structural alterations affect the PK cleavage pattern, it can be challenging to distinguish between binding sites and other structural changes based on LiP-MS results alone.
References
[1] Feng F, Zhang W, Chai Y, Guo D, Chen X. Label-free target protein characterization for small molecule drugs: recent advances in methods and applications. J Pharm Biomed Anal. 2023 Jan 20;223:115107. doi: 10.1016/j.jpba.2022.115107. Epub 2022 Oct 19. PMID: 36334421.
[2] Reber V, Gstaiger M. Target Deconvolution by Limited Proteolysis Coupled to Mass Spectrometry. Methods Mol Biol. 2023;2706:177-190. doi: 10.1007/978-1-0716-3397-7_13. PMID: 37558949.
[3] Malinovska L, Cappelletti V, Kohler D, Piazza I, Tsai TH, Pepelnjak M, Stalder P, Dörig C, Sesterhenn F, Elsässer F, Kralickova L, Beaton N, Reiter L, de Souza N, Vitek O, Picotti P. Proteome-wide structural changes measured with limited proteolysis-mass spectrometry: an advanced protocol for high-throughput applications. Nat Protoc. 2023 Mar;18(3):659-682. doi: 10.1038/s41596-022-00771-x. Epub 2022 Dec 16. Erratum in: Nat Protoc. 2023 Jan 18;: PMID: 36526727.
[4] Kim D, Lee MS, Kim ND, Lee S, Lee HS. Identification of α-amanitin effector proteins in hepatocytes by limited proteolysis-coupled mass spectrometry. Chem Biol Interact. 2023 Dec 1;386:110778. doi: 10.1016/j.cbi.2023.110778. Epub 2023 Oct 23. PMID: 37879594.
[5] Sztacho M, Šalovská B, Červenka J, Balaban C, Hoboth P, Hozák P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells. 2021 Jan 4;10(1):68. doi: 10.3390/cells10010068. PMID: 33406800; PMCID: PMC7824793.
[6] Backe SJ, Votra SD, Stokes MP, Sebestyén E, Castelli M, Torielli L, Colombo G, Woodford MR, Mollapour M, Bourboulia D. PhosY-secretome profiling combined with kinase-substrate interaction screening defines active c-Src-driven extracellular signaling. Cell Rep. 2023 Jun 27;42(6):112539. doi: 10.1016/j.celrep.2023.112539. Epub 2023 May 25. PMID: 37243593; PMCID: PMC10569185.
[7] Piazza I, Beaton N, Bruderer R, Knobloch T, Barbisan C, Chandat L, Sudau A, Siepe I, Rinner O, de Souza N, Picotti P, Reiter L. A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat Commun. 2020 Aug 21;11(1):4200. doi: 10.1038/s41467-020-18071-x. PMID: 32826910; PMCID: PMC7442650.
How to order?

